

Systematic characterization of the HOXA9 downstream targets in MLL-r leukemia by noncoding CRISPR screens
- PMID: 38016946
- PMCID: PMC10684515
- DOI: 10.1038/s41467-023-43264-5
Systematic characterization of the HOXA9 downstream targets in MLL-r leukemia by noncoding CRISPR screens
Abstract
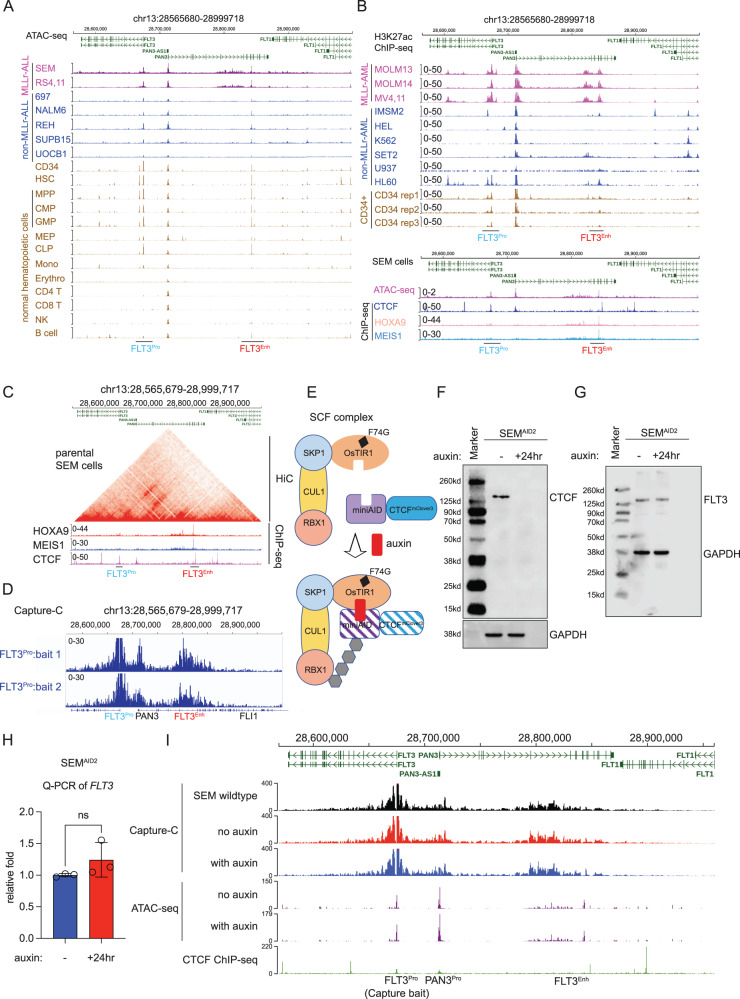
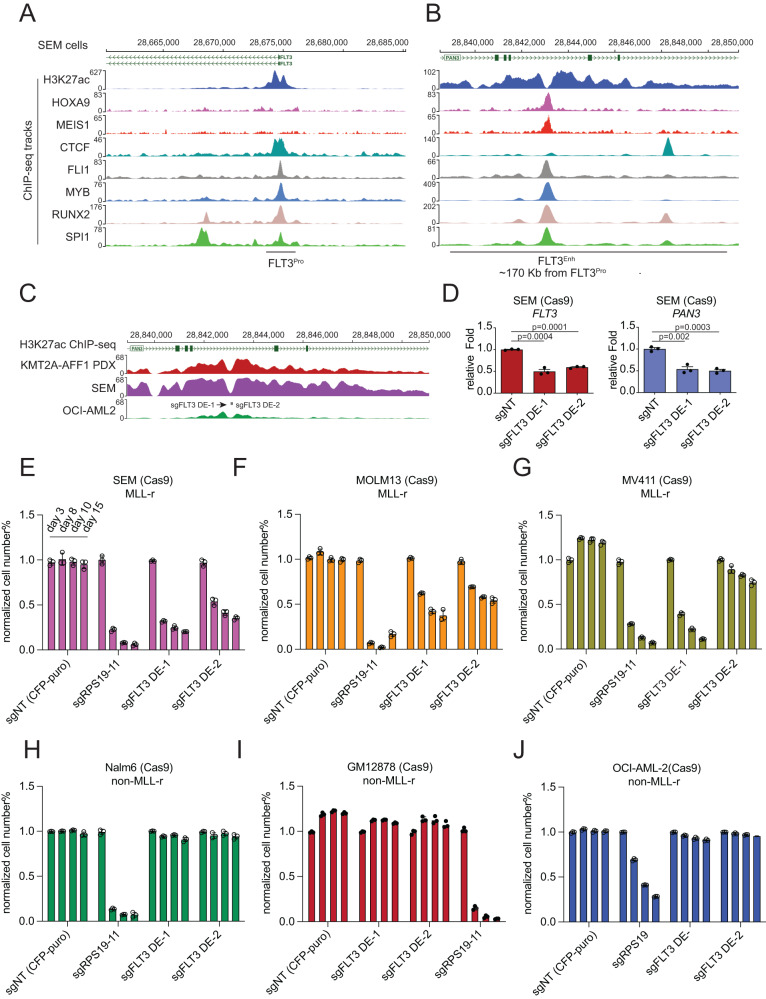
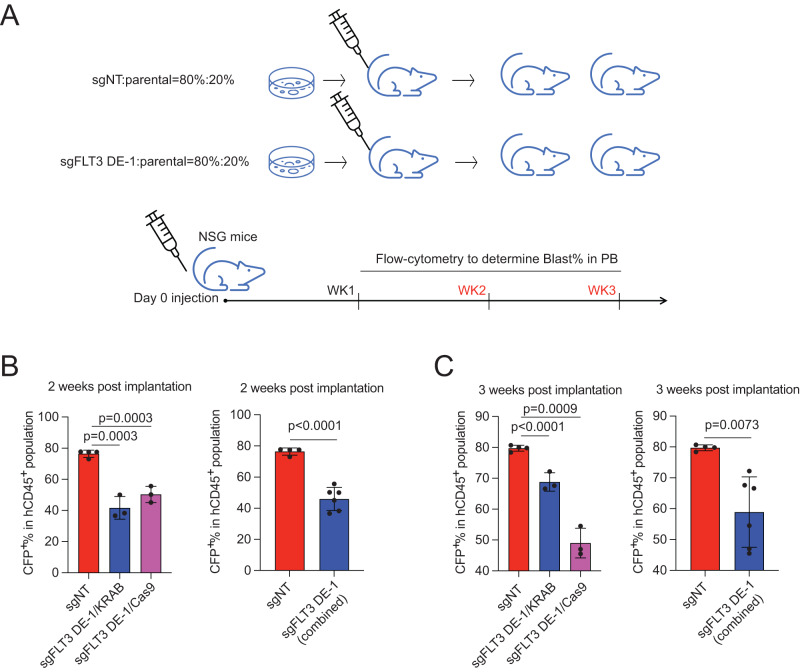
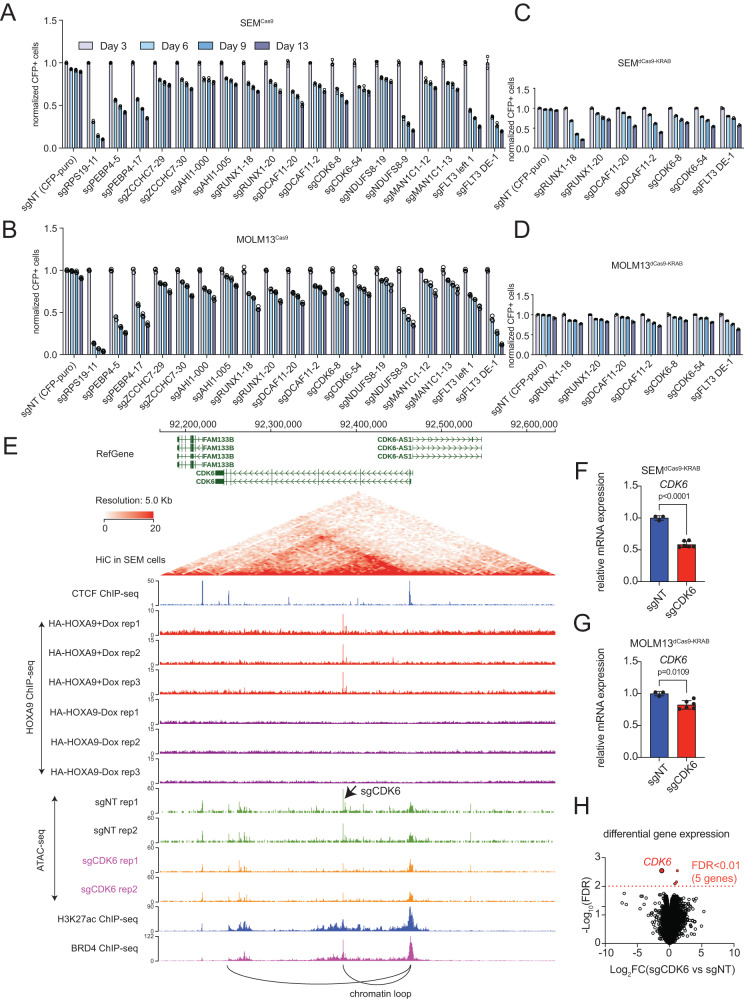
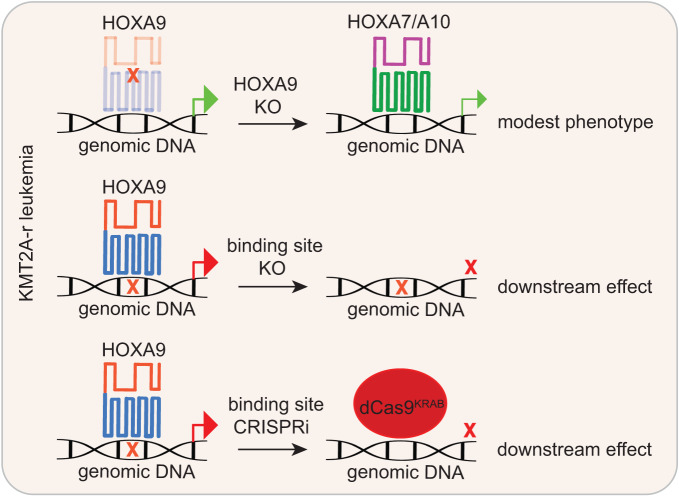
Accumulating evidence indicates that HOXA9 dysregulation is necessary and sufficient for leukemic transformation and maintenance. However, it remains largely unknown how HOXA9, as a homeobox transcriptional factor, binds to noncoding regulatory sequences and controls the downstream genes. Here, we conduct dropout CRISPR screens against 229 HOXA9-bound peaks identified by ChIP-seq. Integrative data analysis identifies reproducible noncoding hits, including those located in the distal enhancer of FLT3 and intron of CDK6. The Cas9-editing and dCas9-KRAB silencing of the HOXA9-bound sites significantly reduce corresponding gene transcription and impair cell proliferation in vitro, and in vivo by transplantation into NSG female mice. In addition, RNA-seq, Q-PCR analysis, chromatin accessibility change, and chromatin conformation evaluation uncover the noncoding regulation mechanism of HOXA9 and its functional downstream genes. In summary, our work improves our understanding of how HOXA9-associated transcription programs reconstruct the regulatory network specifying MLL-r dependency.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
