

Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis
- PMID: 18507500
- PMCID: PMC2429944
- DOI: 10.1371/journal.pmed.0050114
Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis
Abstract
Background: The identification and characterization of tumor suppressor genes has enhanced our understanding of the biology of cancer and enabled the development of new diagnostic and therapeutic modalities. Whereas in past decades, a handful of tumor suppressors have been slowly identified using techniques such as linkage analysis, large-scale sequencing of the cancer genome has enabled the rapid identification of a large number of genes that are mutated in cancer. However, determining which of these many genes play key roles in cancer development has proven challenging. Specifically, recent sequencing of human breast and colon cancers has revealed a large number of somatic gene mutations, but virtually all are heterozygous, occur at low frequency, and are tumor-type specific. We hypothesize that key tumor suppressor genes in cancer may be subject to mutation or hypermethylation.
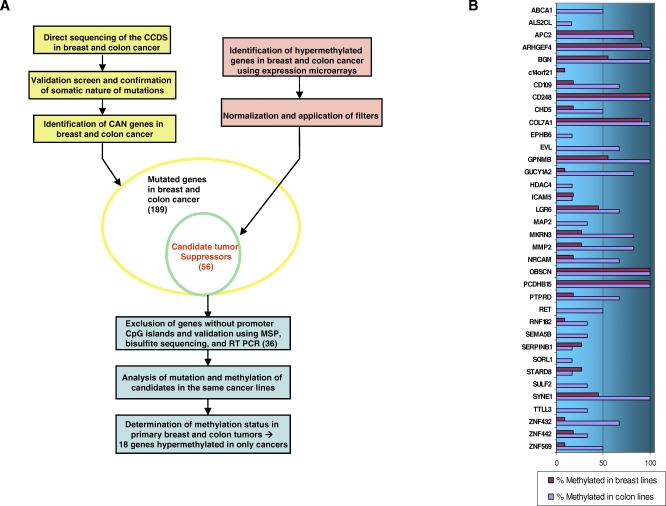
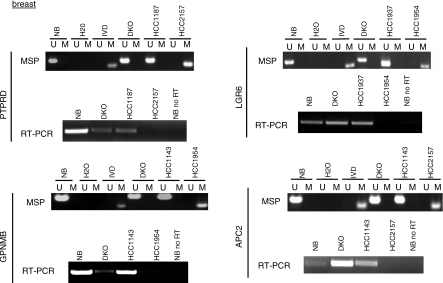
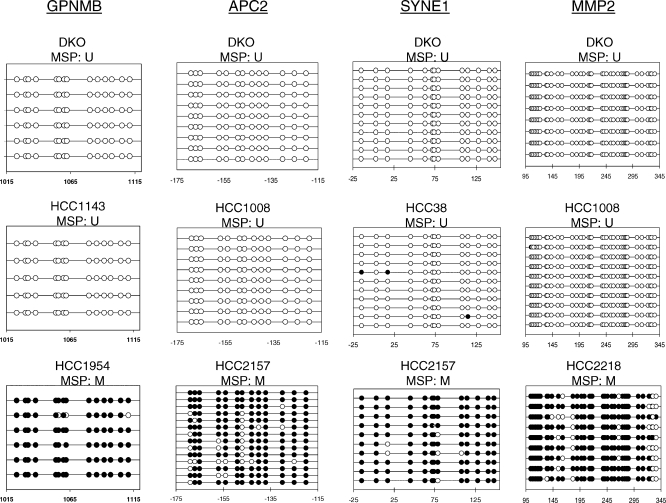
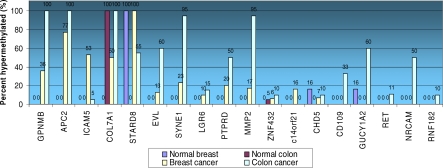
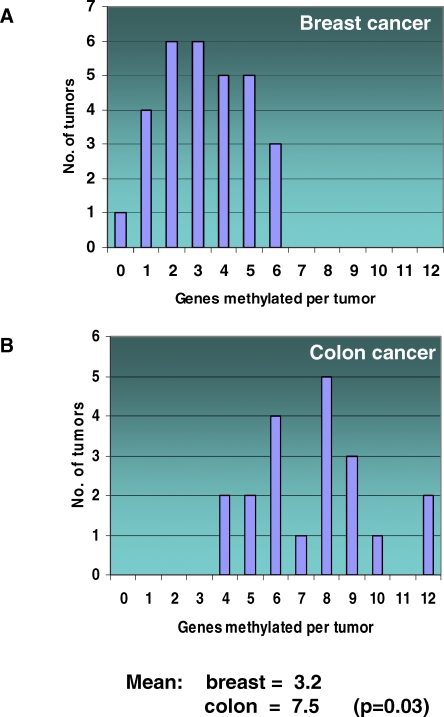
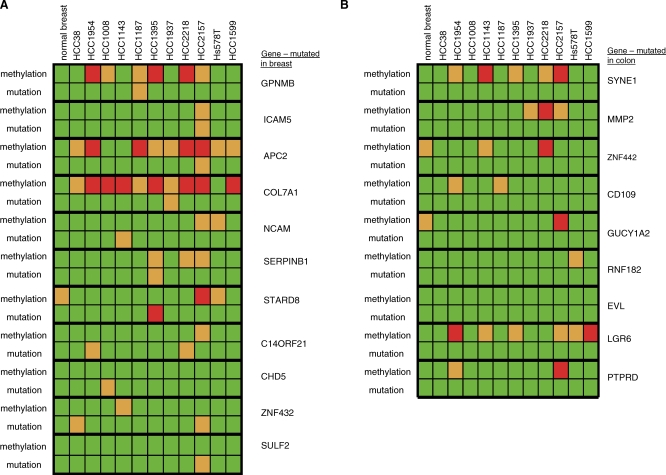
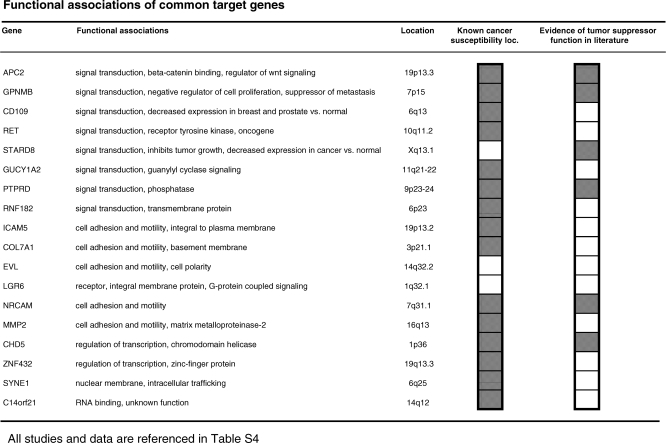
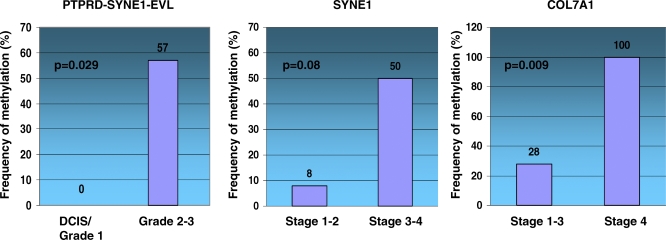
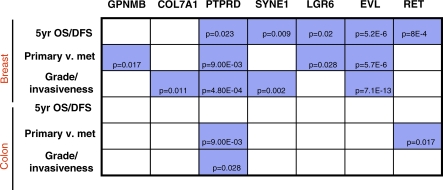
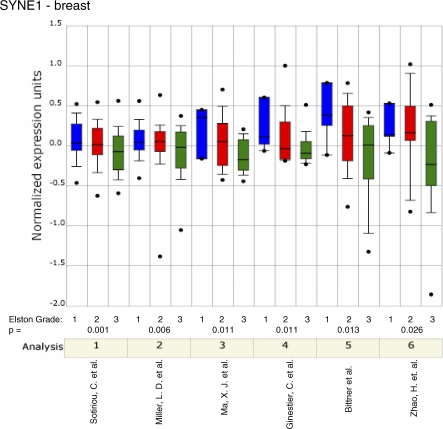
Methods and findings: Here, we show that combined genetic and epigenetic analysis of these genes reveals many with a higher putative tumor suppressor status than would otherwise be appreciated. At least 36 of the 189 genes newly recognized to be mutated are targets of promoter CpG island hypermethylation, often in both colon and breast cancer cell lines. Analyses of primary tumors show that 18 of these genes are hypermethylated strictly in primary cancers and often with an incidence that is much higher than for the mutations and which is not restricted to a single tumor-type. In the identical breast cancer cell lines in which the mutations were identified, hypermethylation is usually, but not always, mutually exclusive from genetic changes for a given tumor, and there is a high incidence of concomitant loss of expression. Sixteen out of 18 (89%) of these genes map to loci deleted in human cancers. Lastly, and most importantly, the reduced expression of a subset of these genes strongly correlates with poor clinical outcome.
Conclusions: Using an unbiased genome-wide approach, our analysis has enabled the discovery of a number of clinically significant genes targeted by multiple modes of inactivation in breast and colon cancer. Importantly, we demonstrate that a subset of these genes predict strongly for poor clinical outcome. Our data define a set of genes that are targeted by both genetic and epigenetic events, predict for clinical prognosis, and are likely fundamentally important for cancer initiation or progression.
Conflict of interest statement
Figures



References
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. - PubMed
-
- Ponder BA. Cancer genetics. Nature. 2001;411:336–341. - PubMed
-
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
