

Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts
- PMID: 24055055
- PMCID: PMC3881975
- DOI: 10.1016/j.celrep.2013.08.022
Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts
Abstract
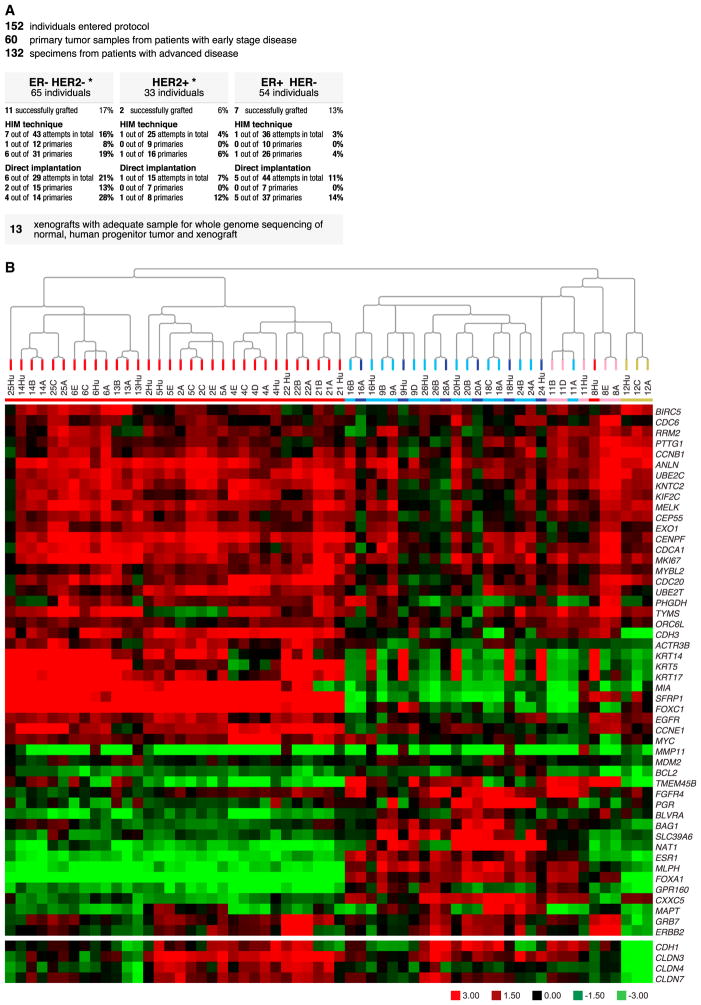
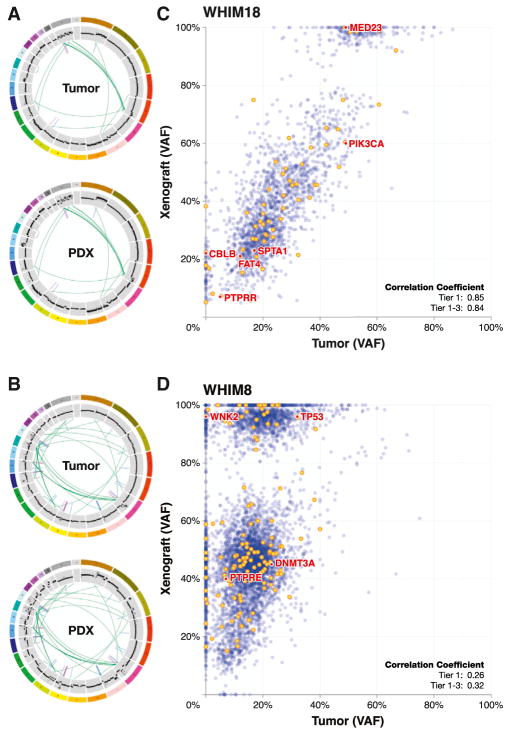
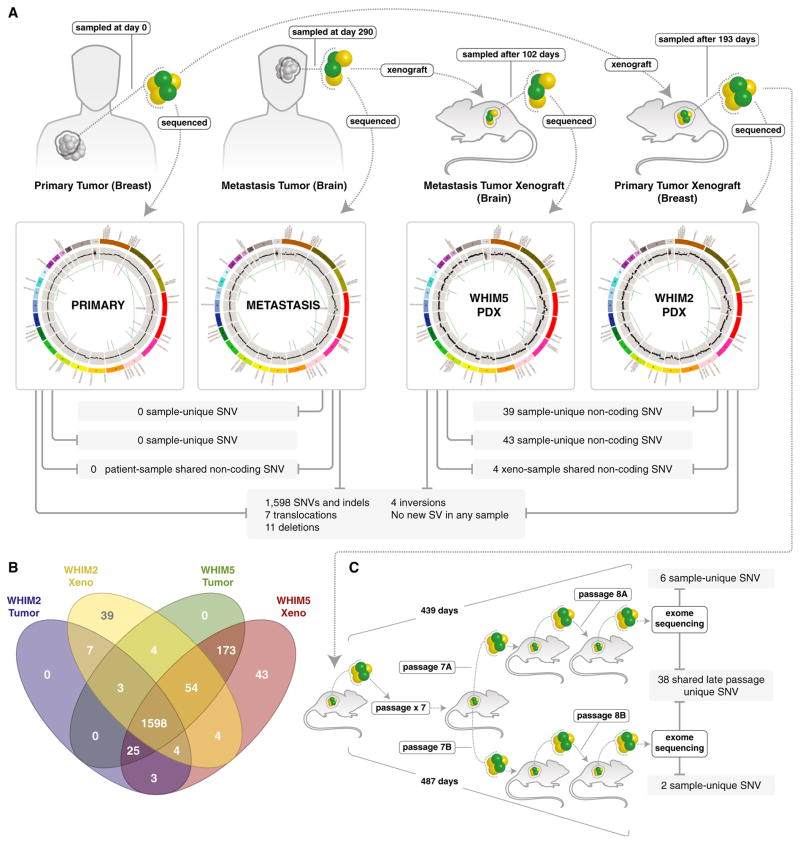
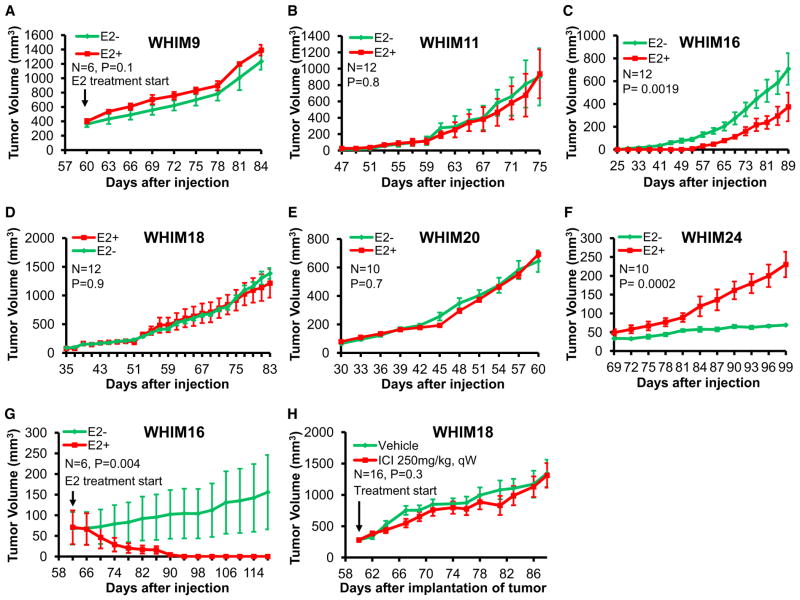
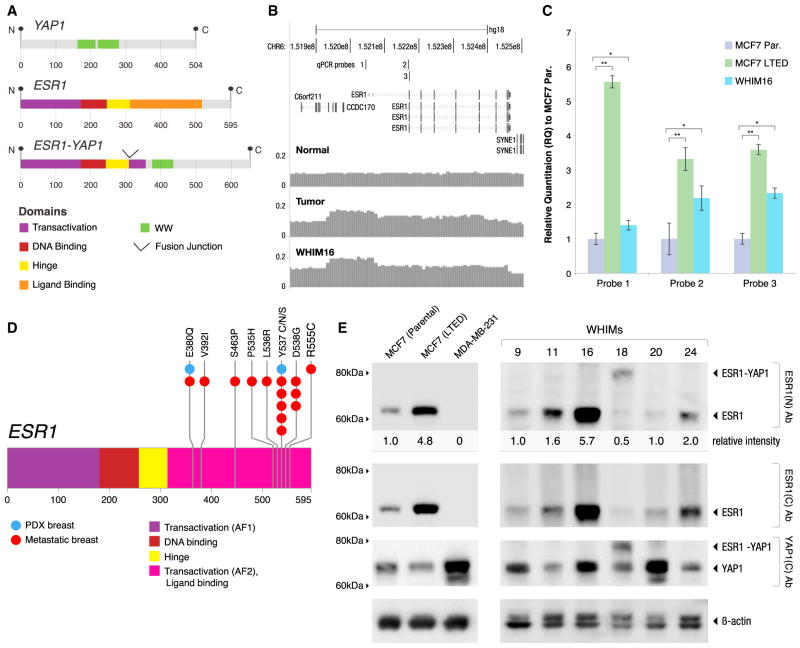
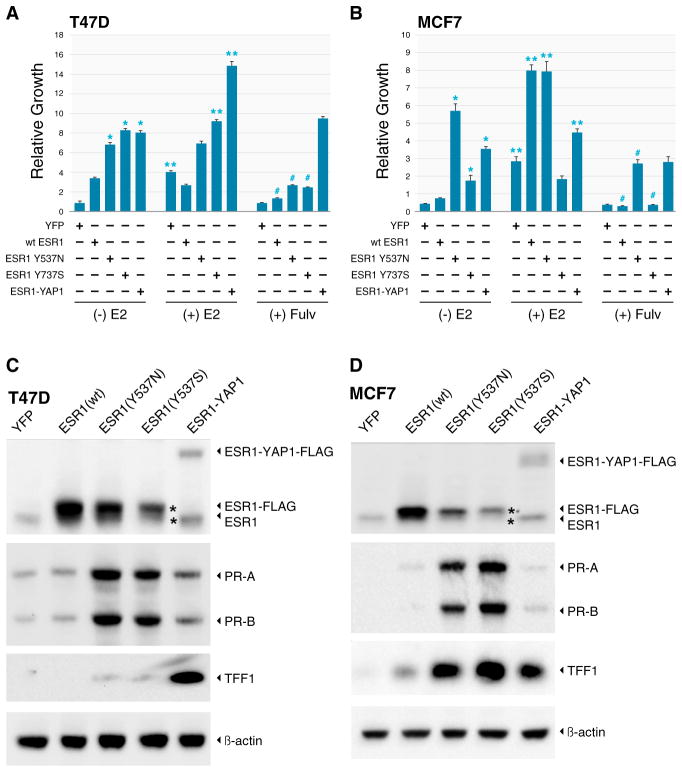
To characterize patient-derived xenografts (PDXs) for functional studies, we made whole-genome comparisons with originating breast cancers representative of the major intrinsic subtypes. Structural and copy number aberrations were found to be retained with high fidelity. However, at the single-nucleotide level, variable numbers of PDX-specific somatic events were documented, although they were only rarely functionally significant. Variant allele frequencies were often preserved in the PDXs, demonstrating that clonal representation can be transplantable. Estrogen-receptor-positive PDXs were associated with ESR1 ligand-binding-domain mutations, gene amplification, or an ESR1/YAP1 translocation. These events produced different endocrine-therapy-response phenotypes in human, cell line, and PDX endocrine-response studies. Hence, deeply sequenced PDX models are an important resource for the search for genome-forward treatment options and capture endocrine-drug-resistance etiologies that are not observed in standard cell lines. The originating tumor genome provides a benchmark for assessing genetic drift and clonal representation after transplantation.
Copyright © 2013 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Using mice to treat (wo)men: mining genetic changes in patient xenografts to attack breast cancer.Cell Rep. 2013 Sep 26;4(6):1061-2. doi: 10.1016/j.celrep.2013.09.008. Cell Rep. 2013. PMID: 24075202
References
-
- Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, et al. In situ analyses of genome instability in breast cancer. Nat Genet. 2004;36:984–988. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
- U24-CA143848/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- P50-CA58223-09A1/CA/NCI NIH HHS/United States
- P50 CA068438/CA/NCI NIH HHS/United States
- U54CA112970/CA/NCI NIH HHS/United States
- P01CA099031/CA/NCI NIH HHS/United States
- P50 CA058223/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- R00 CA127360/CA/NCI NIH HHS/United States
- P01 CA099031/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- UL1 RR024992/RR/NCRR NIH HHS/United States
- U54 CA112970/CA/NCI NIH HHS/United States
- P30 CA16672/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- UL1 TR000448/TR/NCATS NIH HHS/United States
- 3P50 CA68438/CA/NCI NIH HHS/United States
- KG081694/PHS HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
