

Aggressive pituitary tumours and carcinomas, characteristics and management of 171 patients
- PMID: 36018781
- PMCID: PMC9513638
- DOI: 10.1530/EJE-22-0440
Aggressive pituitary tumours and carcinomas, characteristics and management of 171 patients
Abstract
Objective: To describe clinical and pathological characteristics and treatment outcomes in a large cohort of aggressive pituitary tumours (APT)/pituitary carcinomas (PC).
Design: Electronic survey August 2020-May 2021.
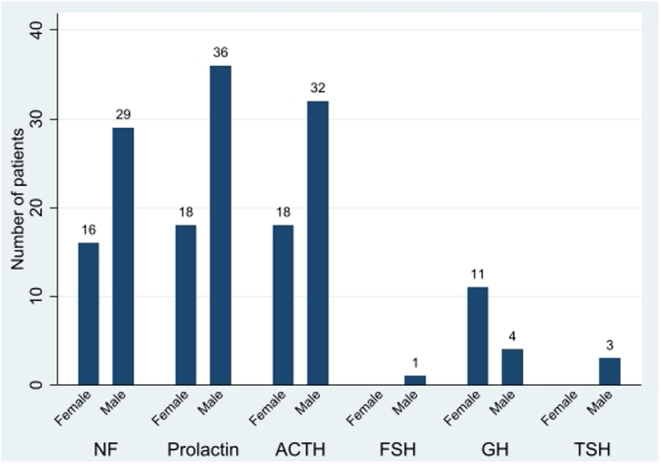
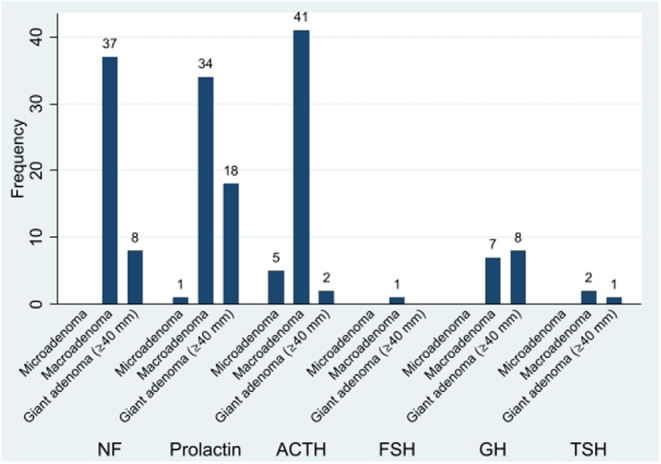
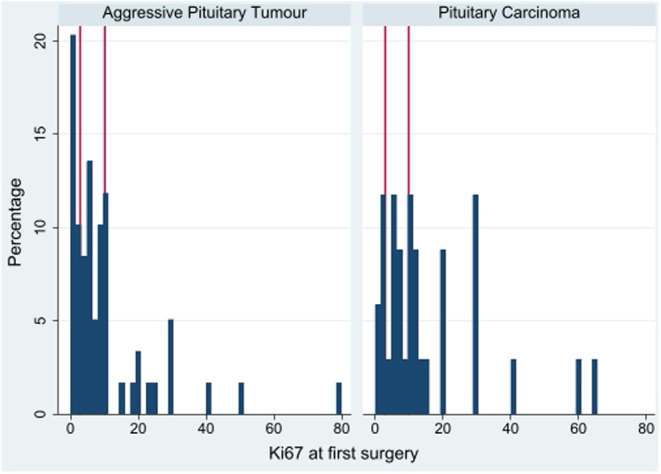
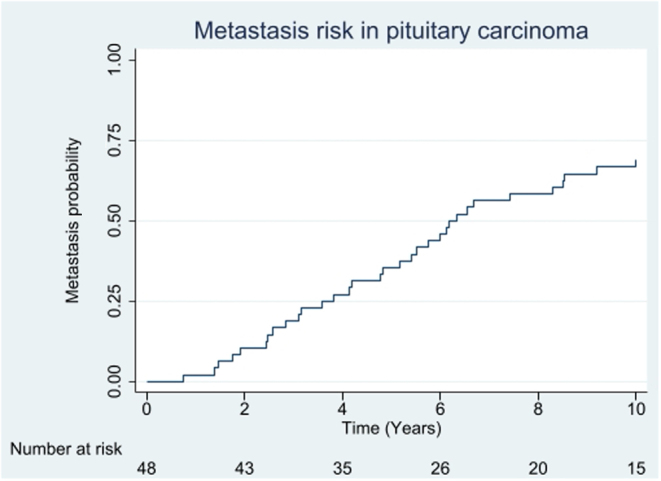
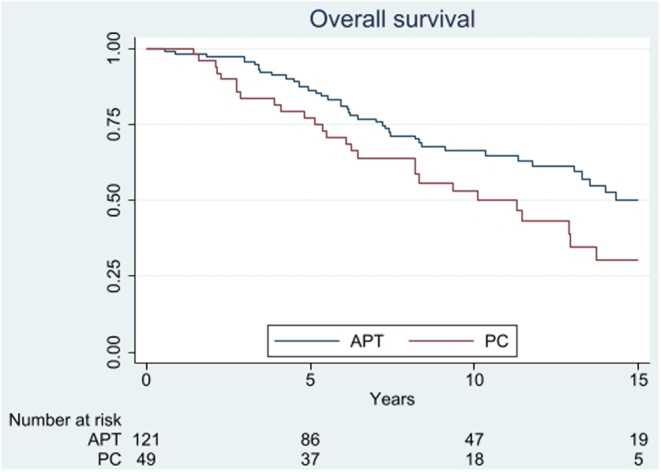
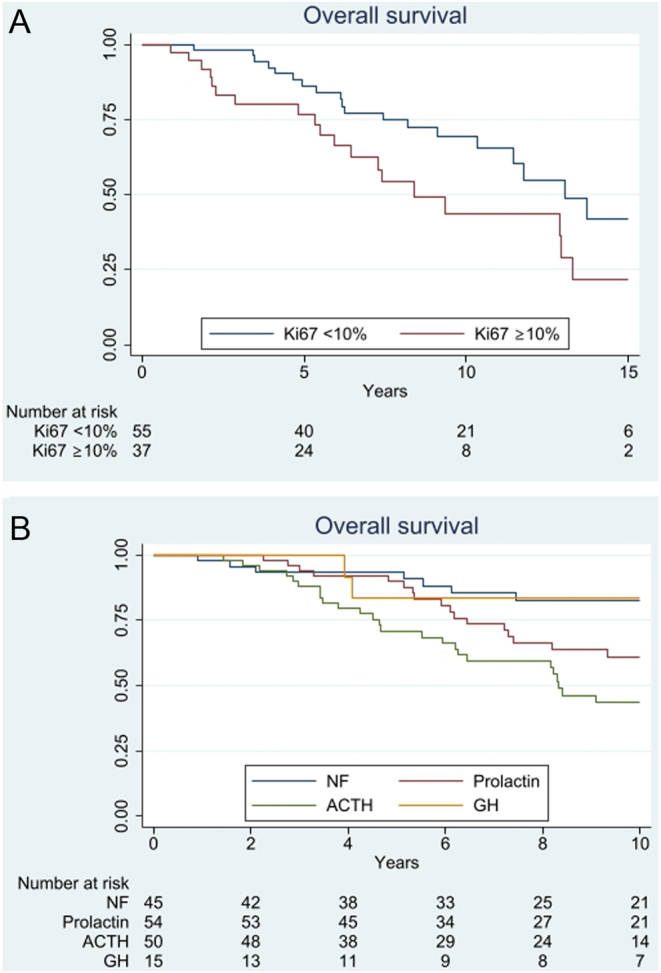
Results: 96% of 171 (121 APT, 50 PC), initially presented as macro/giant tumours, 6 were microadenomas (5 corticotroph). Ninety-seven tumours, initially considered clinically benign, demonstrated aggressive behaviour after 5.5 years (IQR: 2.8-12). Of the patients, 63% were men. Adrenocorticotrophic hormone (ACTH)-secreting tumours constituted 30% of the APT/PC, and the gonadotroph subtypes were under-represented. Five out of 13 silent corticotroph tumours and 2/6 silent somatotroph tumours became secreting. Metastases were observed after median 6.3 years (IQR 3.7-12.1) from diagnosis. At the first surgery, the Ki67 index was ≥3% in 74/93 (80%) and ≥10% in 38/93 (41%) tumours. An absolute increase of Ki67 ≥ 10% after median of 6 years from the first surgery occurred in 18/49 examined tumours. Tumours with an aggressive course from outset had higher Ki67, mitotic counts, and p53. Temozolomide treatment in 156/171 patients resulted in complete response in 9.6%, partial response in 30.1%, stable disease in 28.1%, and progressive disease in 32.2% of the patients. Treatment with bevacizumab, immune checkpoint inhibitors, and peptide receptor radionuclide therapy resulted in partial regression in 1/10, 1/6, and 3/11, respectively. Median survival in APT and PC was 17.2 and 11.3 years, respectively. Tumours with Ki67 ≥ 10% and ACTH-secretion were associated with worse prognosis.
Conclusion: APT/PCs exhibit a wide and challenging spectrum of behaviour. Temozolomide is the first-line chemotherapy, and other oncological therapies are emerging. Treatment response continues to be difficult to predict with currently studied biomarkers.
Figures
References
-
- Roncaroli F, Kovacs K, Lloyd RV, Matsuno A, Righi A. Pituitary carcinoma. In WHO Classification of Tumours of Endocrine Organs, Chapter 1, pp. 41–44. Eds Lloyd RV, Osamura RY, Klöpel G, Rosai J. Lyon, France: Tumours of Pituitary Gland,IARC, 2017.
-
- McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, Burman P. & ESE survey collaborators. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. European Journal of Endocrinology 2018178265–276. (10.1530/EJE-17-0933) - DOI - PubMed
-
- Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, Trouillas J, Dekkers OM. The European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. European Journal of Endocrinology 2018178G1–G24. (10.1530/EJE-17-0796) - DOI - PubMed
-
- Reincke M, Albani A, Assie G, Bancos I, Brue T, Buchfelder M, Chabre O, Ceccato F, Daniele A, Detomas M.et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. European Journal of Endocrinology 2021184P1–P16. (10.1530/EJE-20-1088) - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
