

Identifying recombination hot spots in the HIV-1 genome
- PMID: 24371048
- PMCID: PMC3958072
- DOI: 10.1128/JVI.03014-13
Identifying recombination hot spots in the HIV-1 genome
Abstract
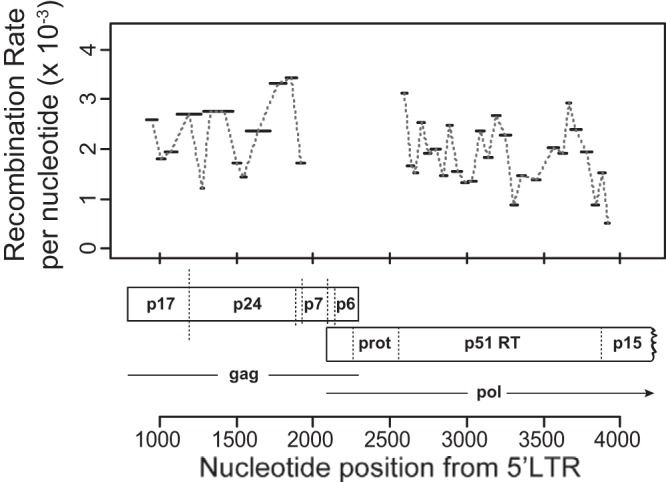
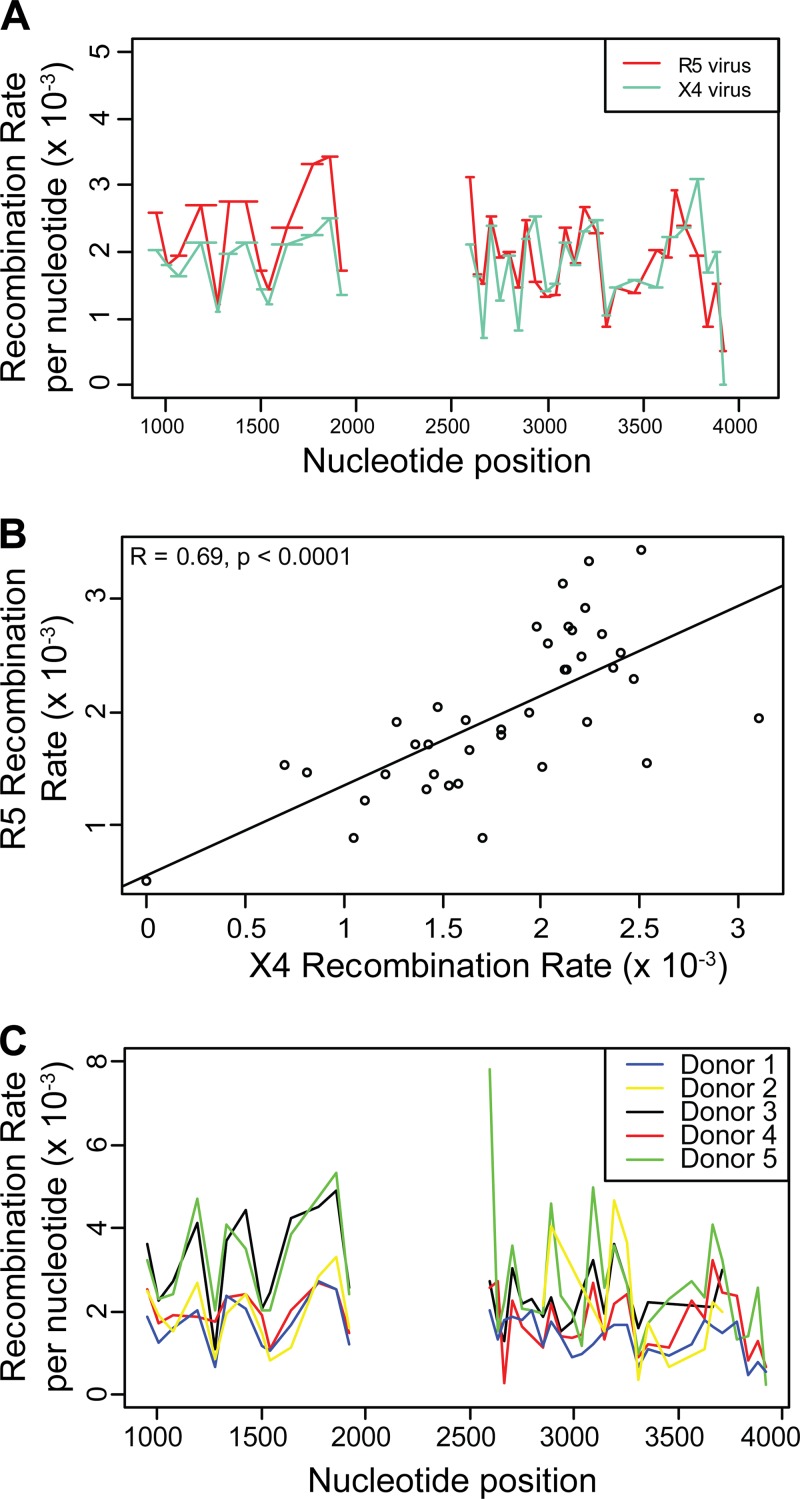
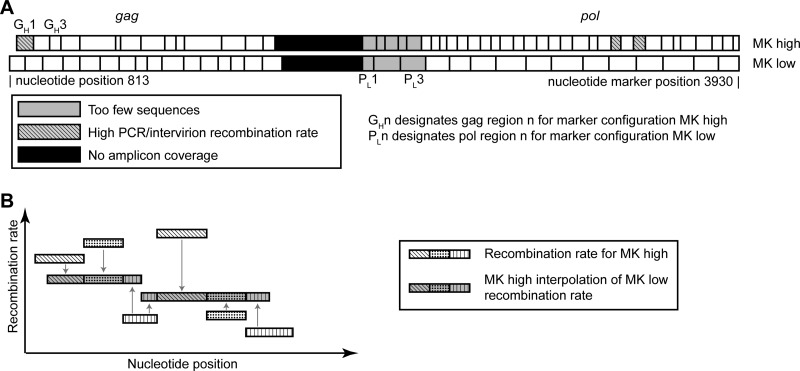
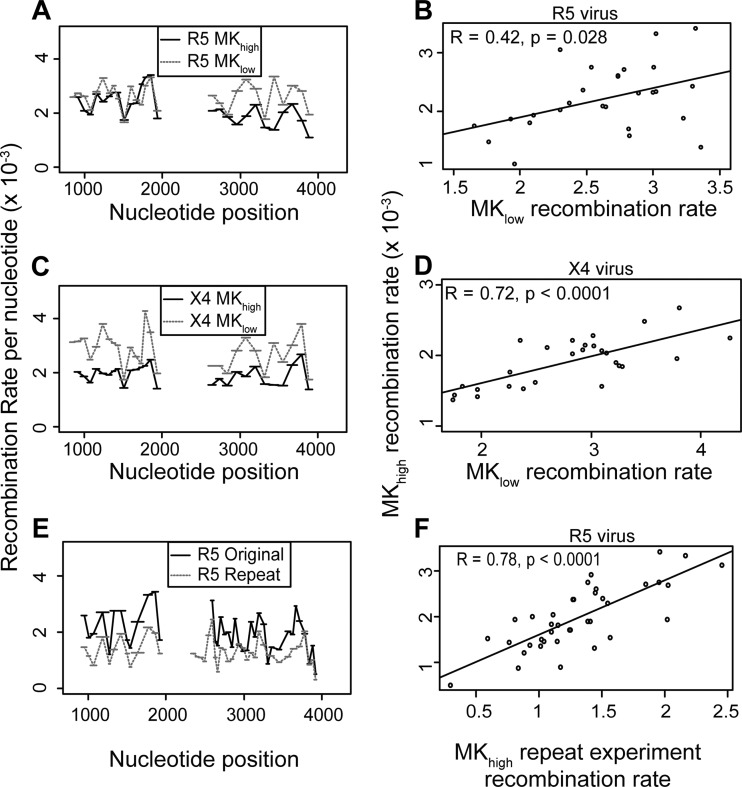
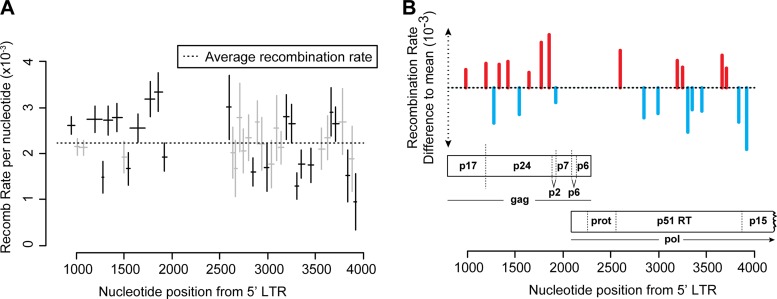
HIV-1 infection is characterized by the rapid generation of genetic diversity that facilitates viral escape from immune selection and antiretroviral therapy. Despite recombination's crucial role in viral diversity and evolution, little is known about the genomic factors that influence recombination between highly similar genomes. In this study, we use a minimally modified full-length HIV-1 genome and high-throughput sequence analysis to study recombination in gag and pol in T cells. We find that recombination is favored at a number of recombination hot spots, where recombination occurs six times more frequently than at corresponding cold spots. Interestingly, these hot spots occur near important features of the HIV-1 genome but do not occur at sites immediately around protease inhibitor or reverse transcriptase inhibitor drug resistance mutations. We show that the recombination hot and cold spots are consistent across five blood donors and are independent of coreceptor-mediated entry. Finally, we check common experimental confounders and find that these are not driving the location of recombination hot spots. This is the first study to identify the location of recombination hot spots between two similar viral genomes with great statistical power and under conditions that closely reflect natural recombination events among HIV-1 quasispecies.
Importance: The ability of HIV-1 to evade the immune system and antiretroviral therapy depends on genetic diversity within the viral quasispecies. Retroviral recombination is an important mechanism that helps to generate and maintain this genetic diversity, but little is known about how recombination rates vary within the HIV-1 genome. We measured recombination rates in gag and pol and identified recombination hot and cold spots, demonstrating that recombination is not random but depends on the underlying gene sequence. The strength and location of these recombination hot and cold spots can be used to improve models of viral dynamics and evolution, which will be useful for the design of robust antiretroviral therapies.
Figures
References
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
