

The combination of whole-exome sequencing and clinical analysis allows better diagnosis of rare syndromic retinal dystrophies
- PMID: 30925032
- PMCID: PMC11377105
- DOI: 10.1111/aos.14095
The combination of whole-exome sequencing and clinical analysis allows better diagnosis of rare syndromic retinal dystrophies
Abstract
Purpose: To identify the accurate clinical diagnosis of rare syndromic inherited retinal diseases (IRDs) based on the combination of clinical and genetic analyses.
Methods: Four unrelated families with various autosomal recessive syndromic inherited retinal diseases were genetically investigated using whole-exome sequencing (WES).
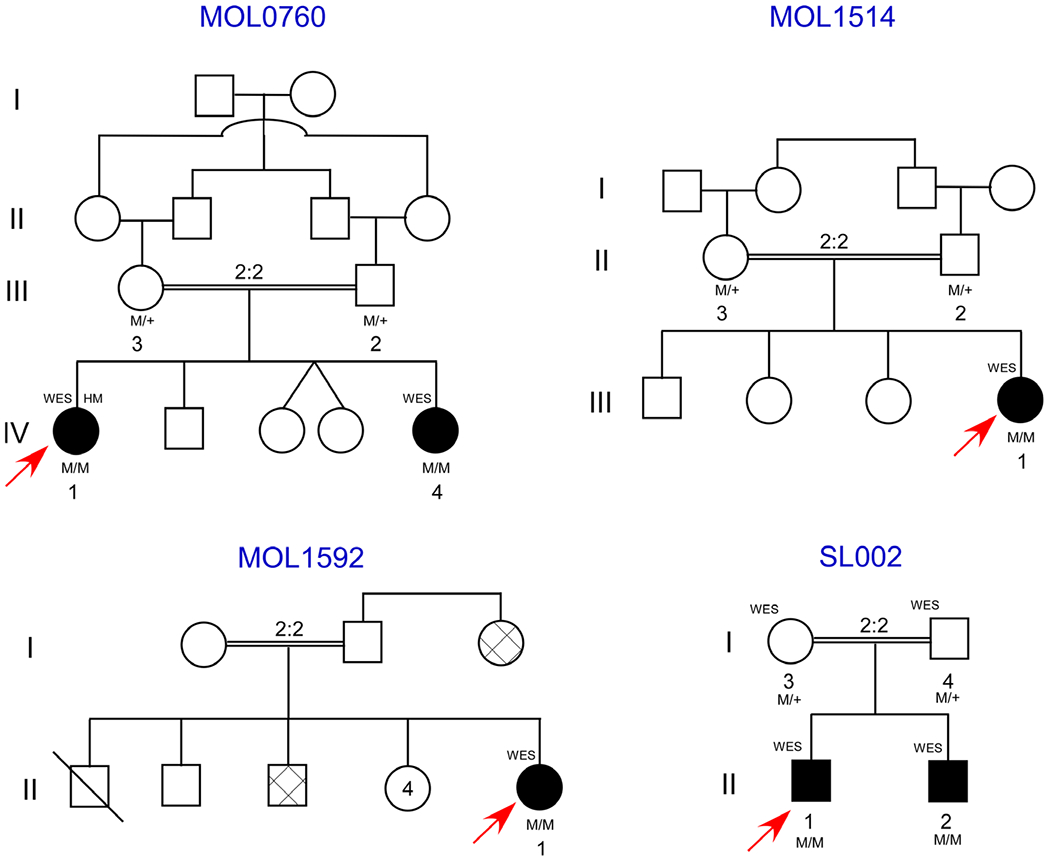
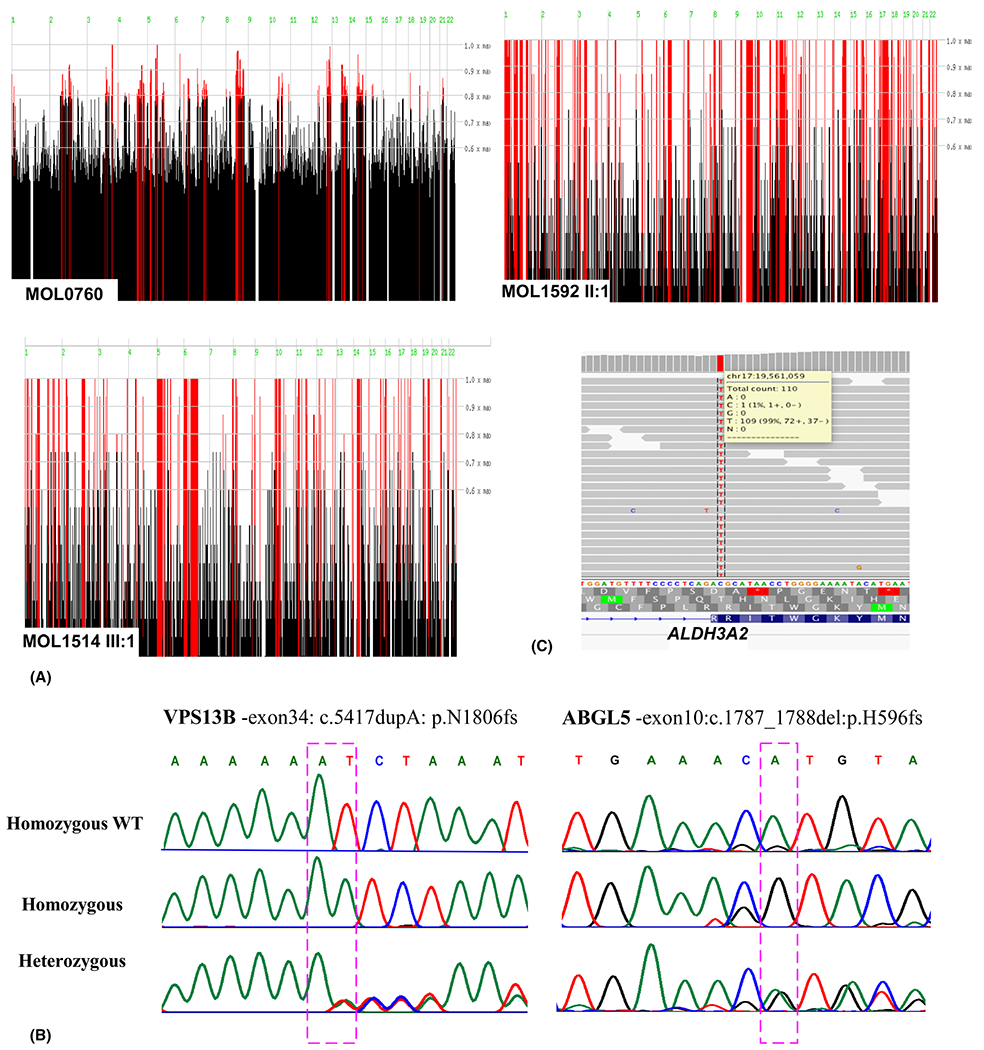
Results: Two affected subjects in family MOL0760 presented with a distinctive combination of short stature, developmental delay, congenital mental retardation, microcephaly, facial dysmorphism and retinitis pigmentosa (RP). Subjects were clinically diagnosed with suspected Kabuki syndrome. WES revealed a homozygous nonsense mutation (c.5492dup, p.Asn1831Lysfs*8) in VPS13B that is known to cause Cohen syndrome. The index case of family MOL1514 presented with both RP and liver dysfunction, suspected initially to be related. WES identified a homozygous frameshift mutation (c.1787_1788del, p.His596Argfs*47) in AGBL5, associated with nonsyndromic RP. The MOL1592 family included three affected subjects with crystalline retinopathy, skin ichthyosis, short stature and congenital adrenal hypoplasia, and were found to harbour a homozygous nonsense mutation (c.682C>T, p.Arg228Cys) in ALDH3A2, reported to cause Sjögren-Larsson syndrome (SLS). In the fourth family, SJ002, two siblings presented with hypotony, psychomotor delay, dysmorphic facial features, pathologic myopia, progressive external ophthalmoplegia and diffuse retinal atrophy. Probands were suspected to have atypical Kearns-Sayre syndrome, but were diagnosed with combined oxidative phosphorylation deficiency-20 due to a novel suspected missense variant (c.1691C>T, p.Ala564Val) in VARS2.
Conclusion: Our findings emphasize the important complement of WES and thorough clinical investigation in establishing precise clinical diagnosis. This approach constitutes the basis for personalized medicine in rare IRDs.
Keywords: inherited rare disease; precise clinical diagnosis; syndromic retinal disease; whole-exome sequencing.
© 2019 Acta Ophthalmologica Scandinavica Foundation. Published by John Wiley & Sons Ltd.
Figures

References
-
- Astuti GDN, Arno G, Hull S et al. (2016): Mutations in AGBL5, encoding a-tubulin deglutamylase, are associated with autosomal recessive retinitis pigmentosa. Investig Ophthalmol Vis Sci 57: 6180–6187. - PubMed
-
- Bacchelli C & Williams HJ (2016): Opportunities and technical challenges in next-generation sequencing for diagnosis of rare pediatric diseases. Expert Rev Mol Diagn 16: 1073–1082. - PubMed
-
- Baertling F, Alhaddad B, Seibt A et al. (2017): Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metab Brain Dis 32: 267–270. - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
