

A genomewide linkage-disequilibrium scan localizes the Saguenay-Lac-Saint-Jean cytochrome oxidase deficiency to 2p16
- PMID: 11156535
- PMCID: PMC1235273
- DOI: 10.1086/318197
A genomewide linkage-disequilibrium scan localizes the Saguenay-Lac-Saint-Jean cytochrome oxidase deficiency to 2p16
Abstract
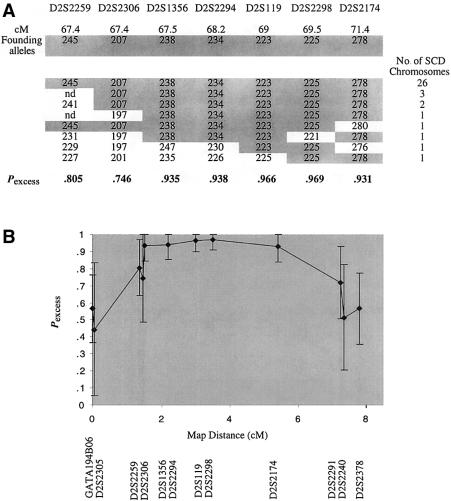
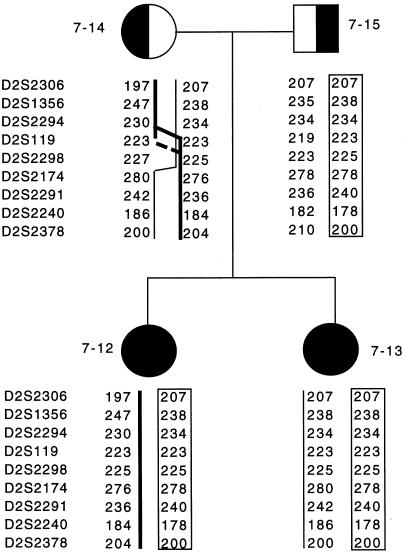
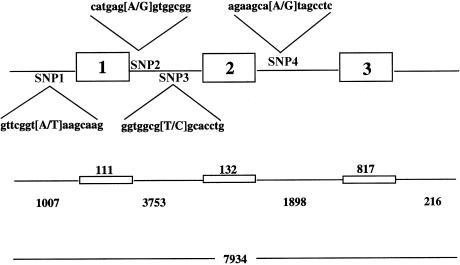
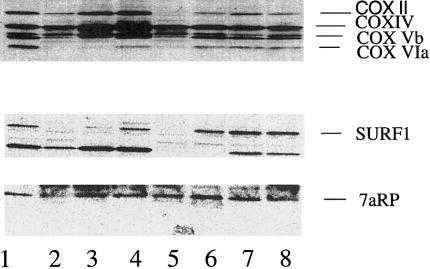
Leigh syndrome (LS) affects 1/40,000 newborn infants in the worldwide population and is characterized by the presence of developmental delay and lactic acidosis and by a mean life expectancy variously estimated at 3-5 years. Saguenay-Lac-Saint-Jean (SLSJ) cytochrome oxidase (COX) deficiency (LS French-Canadian type [LSFC] [MIM 220111]), an autosomal recessive form of congenital lactic acidosis, presents with developmental delay and hypotonia. It is an LS variant that is found in a geographically isolated region of Quebec and that occurs in 1/2,178 live births. Patients with LSFC show a phenotype similar to that of patients with LS, but the two groups differ in clinical presentation. We studied DNA samples from 14 patients with LSFC and from their parents, representing a total of 13 families. Because of founder effects in the SLSJ region, considerable linkage disequilibrium (LD) was expected to surround the LSFC mutation. We therefore performed a genomewide screen for LD, using 290 autosomal microsatellite markers. A single marker, D2S1356, located on 2p16, showed significant (P < 10(-5)) genomewide LD. Using high-resolution genetic mapping with additional markers and four additional families with LSFC, we were able to identify a common ancestral haplotype and to limit the critical region to approximately 2 cM between D2S119 and D2S2174. COX7AR, a gene encoding a COX7a-related protein, had previously been mapped to this region. We determined the genomic structure and resequenced this gene in patients with LSFC and in controls but found no functional mutations. Although the LSFC gene remains to be elucidated, the present study demonstrates the feasibility of using a genomewide LD strategy to localize the critical region for a rare genetic disease in a founder population.
Figures
References
Electronic-Database Information
-
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://www.marshmed.org/genetics
-
- CHLC, http://lpg.nci.nih.gov/CHLC/ (for fluorescence-labeled microsatellite markers)
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for accession number AB007618)
-
- GeneMap'98, http://www.ncbi.nlm.nih.gov/genemap98/ (for cDNA sequence of a possible candidate gene, COX7AR [accession number AB007618])
-
- Généthon, http://www.genethon.fr/genethon_en.html (for additional markers flanking marker D2S1356)
References
-
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Borgeois M, Viegas-Péquignot E, Munnich A, Rötig A (1995) Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 11:144–149 - PubMed
-
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Durr A, Fontaine B, Ballabio A (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93:973–983 - PubMed
-
- Couture P, Morisette J, Gaudet D, Vohl M, Gagne C, Bergeron J, Sepres J-P, Simard J (1999) Fine mapping of low-density lipoprotein receptor gene by genetic linkage on chromosome 19p13.1-p13.3 and study of the founder effect of four French Canadian low-density lipoprotein receptor gene mutations. Atherosclerosis 143:145–151 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
