

Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1
- PMID: 11781871
- PMCID: PMC384941
- DOI: 10.1086/338998
Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1
Abstract
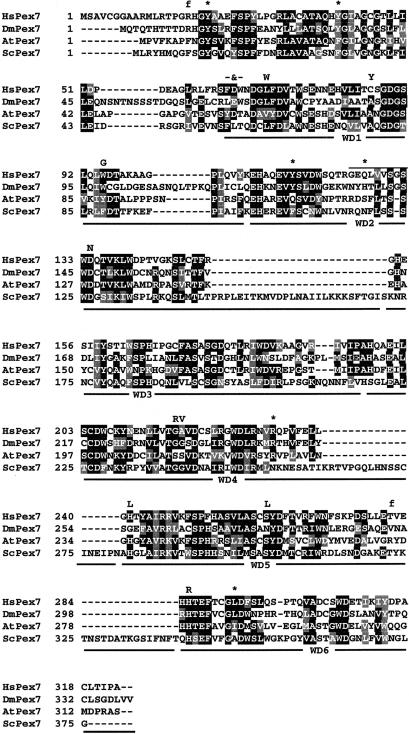
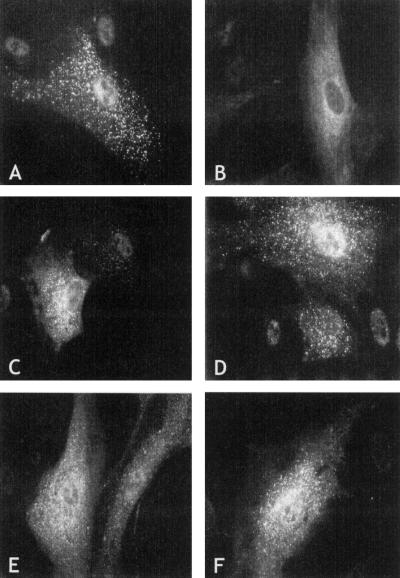
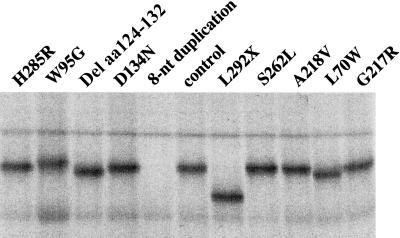
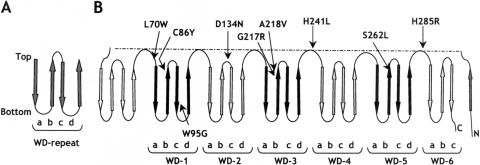
Rhizomelic chondrodysplasia punctata (RCDP) is a genetically heterogeneous, autosomal recessive disorder of peroxisomal metabolism that is clinically characterized by symmetrical shortening of the proximal long bones, cataracts, periarticular calcifications, multiple joint contractures, and psychomotor retardation. Most patients with RCDP have mutations in the PEX7 gene encoding peroxin 7, the cytosolic PTS2-receptor protein required for targeting a subset of enzymes to peroxisomes. These enzymes are deficient in cells of patients with RCDP, because of their mislocalization to the cytoplasm. We report the mutational spectrum in the PEX7 gene of 78 patients (including five pairs of sibs) clinically and biochemically diagnosed with RCDP type I. We found 22 different mutations, including 18 novel ones. Furthermore, we show by functional analysis that disease severity correlates with PEX7 allele activity: expression of eight different alleles from patients with severe RCDP failed to restore the targeting defect in RCDP fibroblasts, whereas two alleles found only in patients with mild disease complemented the targeting defect upon overexpression. Surprisingly, one of the mild alleles comprises a duplication of nucleotides 45-52, which is predicted to lead to a frameshift at codon 17 and an absence of functional peroxin 7. The ability of this allele to complement the targeting defect in RCDP cells suggests that frame restoration occurs, resulting in full-length functional peroxin 7, which leads to amelioration of the predicted severe phenotype. This was confirmed in vitro by expression of the eight-nucleotide duplication-containing sequence fused in different reading frames to the coding sequence of firefly luciferase in COS cells.
Figures
References
Electronic-Database Information
-
- ExPasy Secondary Structure Prediction Package, http://www.expasy.ch/#secondary
-
- Flybase, http://hedgehog.lbl.gov:7081 (for Drosophila melanogaster PEX7 orthologue [annotation number Fban0006486])
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for human PEX7 gene [accession numbers AF180806–AF180814], for PEX7 cDNA [accession number U69171], Arabidopsis thaliana PEX7 orthologue [accession number AF130973], and Saccharomyces cerevisiae PEX7 orthologue [accession number X81424])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RCDP type 1 [MIM 215100], RCDP type 2 [MIM 222765], and RCDP type 3 [MIM 600121])
-
- PHD Program for Protein Structure Prediction, http://www.embl-heidelberg.de/predictprotein/
References
-
- Barth PG, Wanders RJA, Schutgens RBH, Staalman C (1996) Variant RCDP with normal phytanic acid: clinico-biochemical delineation of a subtype and complementation studies. Am J Med Genet 62:164–168 - PubMed
-
- Baumgartner MR, Poll-The BT, Verhoeven NM, Jakobs C, Espeel M, Roels F, Rabier D, Levade T, Rolland MO, Martinez M, Wanders RJA, Saudubray JM (1998) Clinical approach to inherited peroxisomal disorders: a series of 27 patients. Ann Neurol 44:720–730 - PubMed
-
- Braverman N, Steel G, Lin P, Moser A, Moser H, Valle D (2000) PEX7 gene structure, alternative transcripts and evidence for a founder haplotype for the frequent RCDP allele L292ter. Genomics 63:181–192 - PubMed
-
- Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D (1997) Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet 15:369–376 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
