

Shigella applies molecular mimicry to subvert vinculin and invade host cells
- PMID: 17088427
- PMCID: PMC2064523
- DOI: 10.1083/jcb.200605091
Shigella applies molecular mimicry to subvert vinculin and invade host cells
Abstract
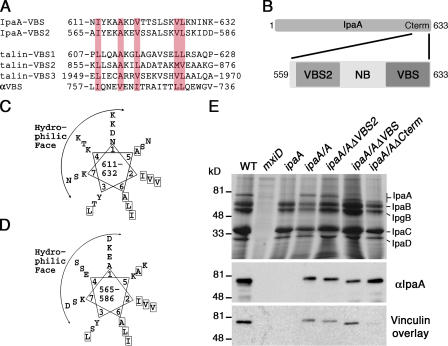
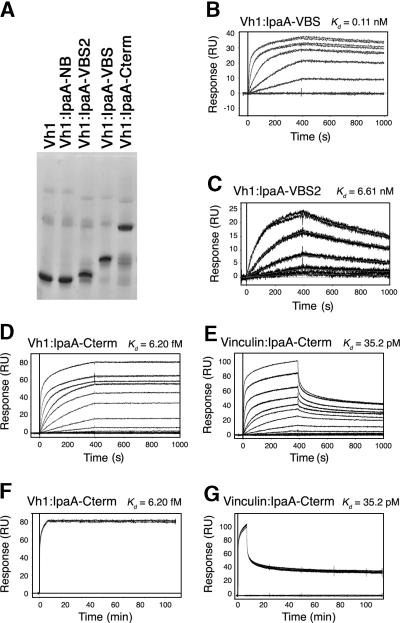
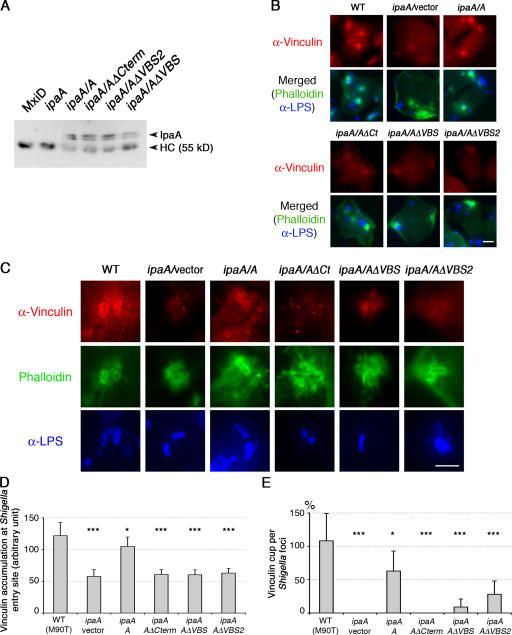
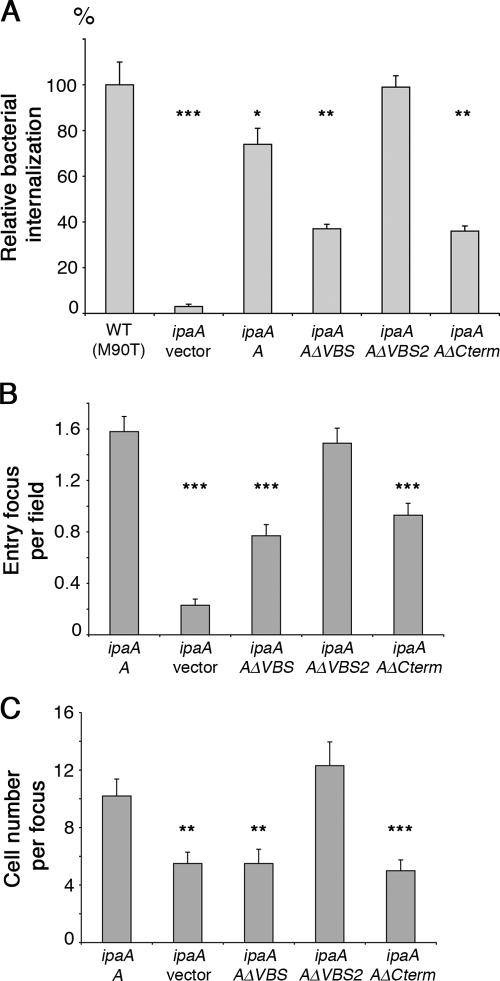
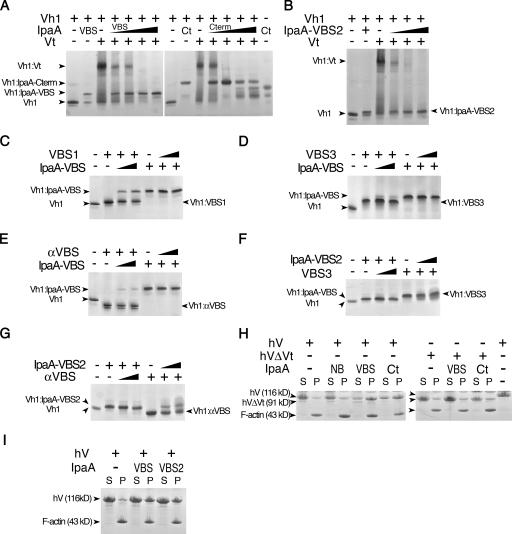
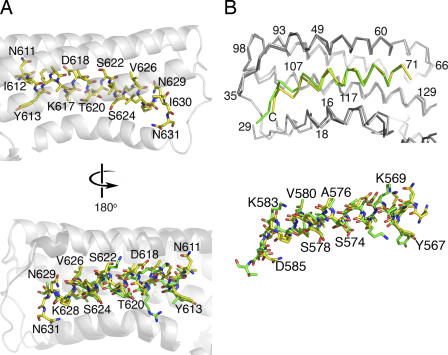
Shigella flexneri, the causative agent of bacillary dysentery, injects invasin proteins through a type III secretion apparatus upon contacting the host cell, which triggers pathogen internalization. The invasin IpaA is essential for S. flexneri pathogenesis and binds to the cytoskeletal protein vinculin to facilitate host cell entry. We report that IpaA harbors two vinculin-binding sites (VBSs) within its C-terminal domain that bind to and activate vinculin in a mutually exclusive fashion. Only the highest affinity C-terminal IpaA VBS is necessary for efficient entry and cell-cell spread of S. flexneri, whereas the lower affinity VBS appears to contribute to vinculin recruitment at entry foci of the pathogen. Finally, the crystal structures of vinculin in complex with the VBSs of IpaA reveal the mechanism by which IpaA subverts vinculin's functions, where S. flexneri utilizes a remarkable level of molecular mimicry of the talin-vinculin interaction to activate vinculin. Mimicry of vinculin's interactions may therefore be a general mechanism applied by pathogens to infect the host cell.
Figures
References
-
- Bakolitsa, C., D.M. Cohen, L.A. Bankston, A.A. Bobkov, G.W. Cadwell, L. Jennings, D.R. Critchley, S.W. Craig, and R.C. Liddington. 2004. Structural basis for vinculin activation at sites of cell adhesion. Nature. 430:583–586. - PubMed
-
- Bakolitsa, C., J.M. de Pereda, C.R. Bagshaw, D.R. Critchley, and R.C. Liddington. 1999. Crystal structure of the vinculin tail suggests a pathway for activation. Cell. 99:603–613. - PubMed
-
- Bois, P.R.J., B.P. O'Hara, D. Nietlispach, J. Kirkpatrick, and T. Izard. 2006. The vinculin binding sites of talin and α-actinin are sufficient to activate vinculin. J. Biol. Chem. 281:7228–7236. - PubMed
-
- Borgon, R.A., C. Vonrhein, G. Bricogne, P.R.J. Bois, and T. Izard. 2004. Crystal structure of human vinculin. Structure. 12:1189–1197. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
