

Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity
- PMID: 17349580
- PMCID: PMC1939942
- DOI: 10.1016/j.ccr.2006.12.017
Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity
Abstract
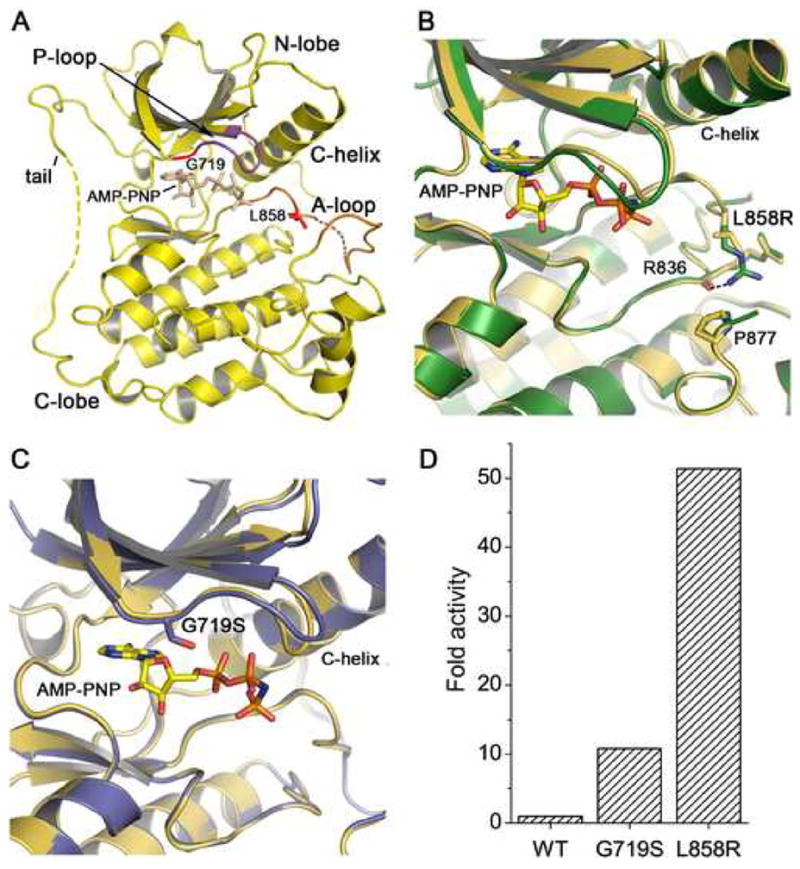
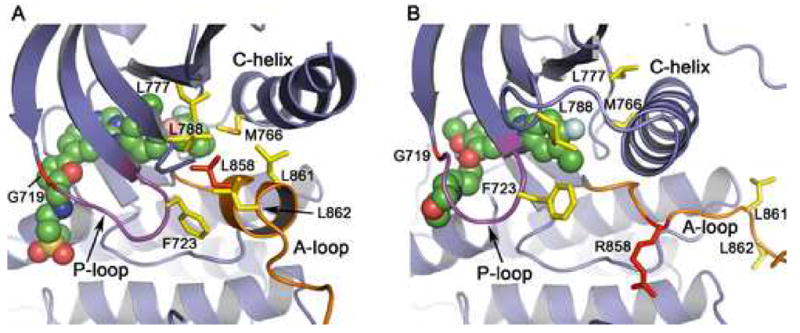
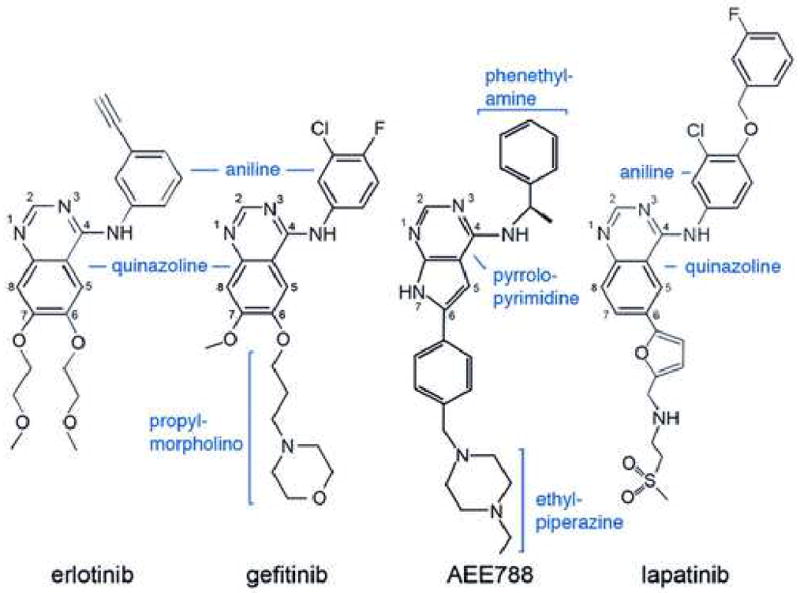
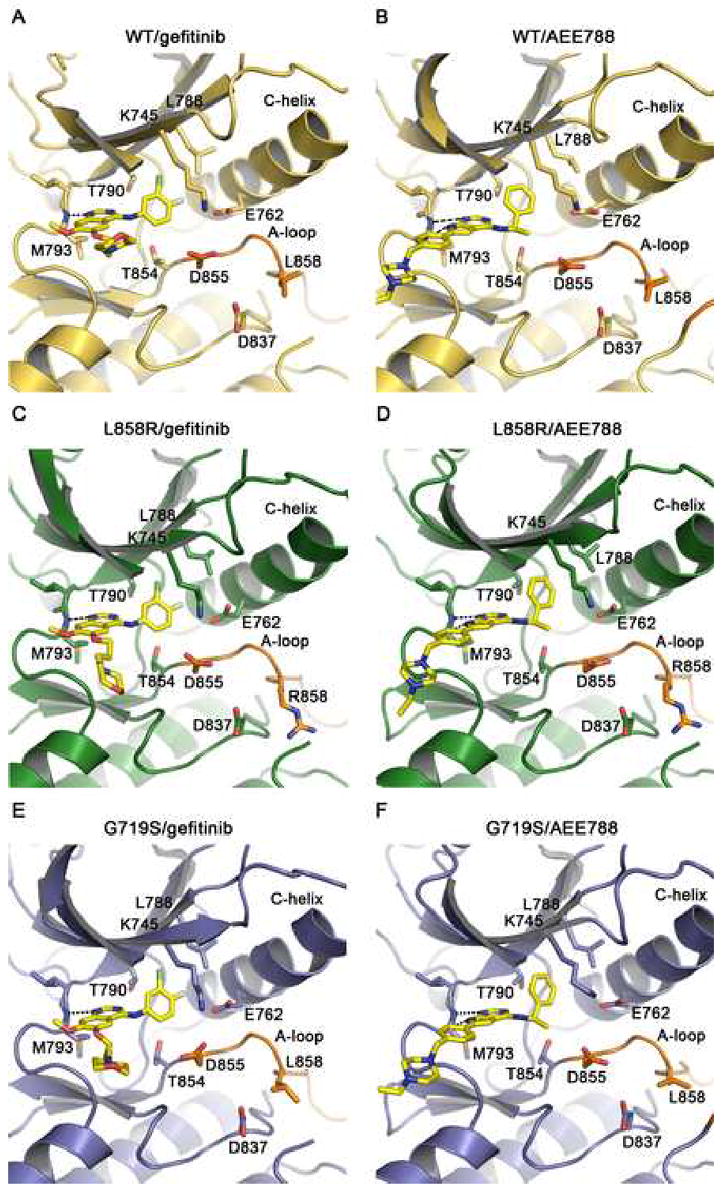
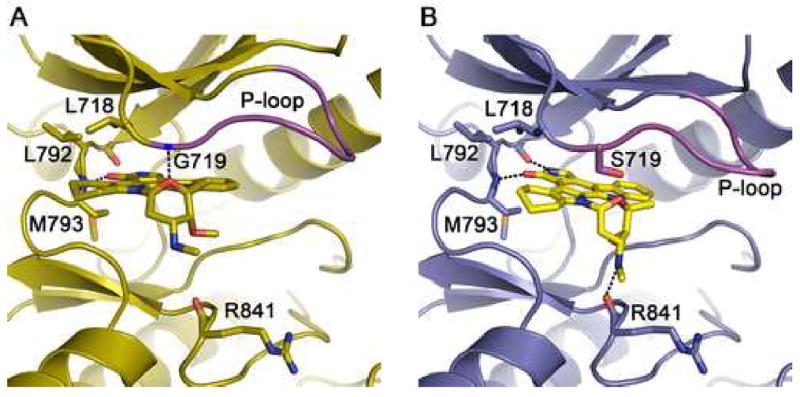
Mutations in the EGFR kinase are a cause of non-small-cell lung cancer. To understand their mechanism of activation and effects on drug binding, we studied the kinetics of the L858R and G719S mutants and determined their crystal structures with inhibitors including gefitinib, AEE788, and a staurosporine. We find that the mutations activate the kinase by disrupting autoinhibitory interactions, and that they accelerate catalysis as much as 50-fold in vitro. Structures of inhibitors in complex with both wild-type and mutant kinases reveal similar binding modes for gefitinib and AEE788, but a marked rotation of the staurosporine in the G719S mutant. Strikingly, direct binding measurements show that gefitinib binds 20-fold more tightly to the L858R mutant than to the wild-type enzyme.
Figures
Comment in
-
The EGF receptor Hokey-Cokey.Cancer Cell. 2007 Mar;11(3):209-11. doi: 10.1016/j.ccr.2007.02.021. Cancer Cell. 2007. PMID: 17349577
References
-
- Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart SJ, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. - PubMed
-
- Arao T, Fukumoto H, Takeda M, Tamura T, Saijo N, Nishio K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004;64:9101–9104. - PubMed
-
- Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Henthorn L, Richards L. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–1914. - PubMed
-
- Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, Knight WB. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry. 1995;34:14843–14851. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
