

Exploring the repeat protein universe through computational protein design
- PMID: 26675729
- PMCID: PMC4845728
- DOI: 10.1038/nature16162
Exploring the repeat protein universe through computational protein design
Abstract
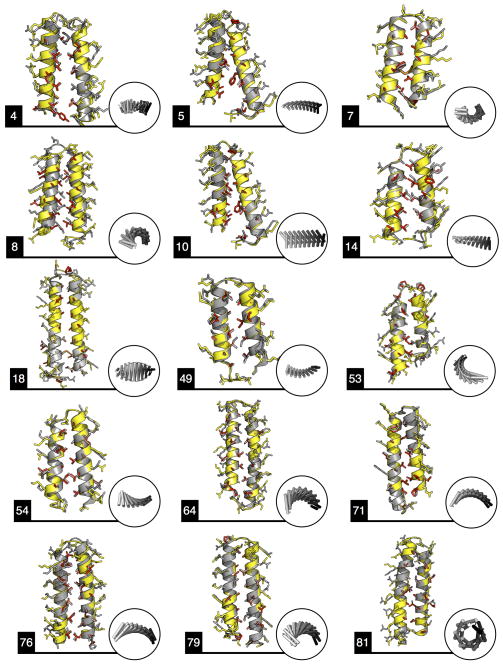
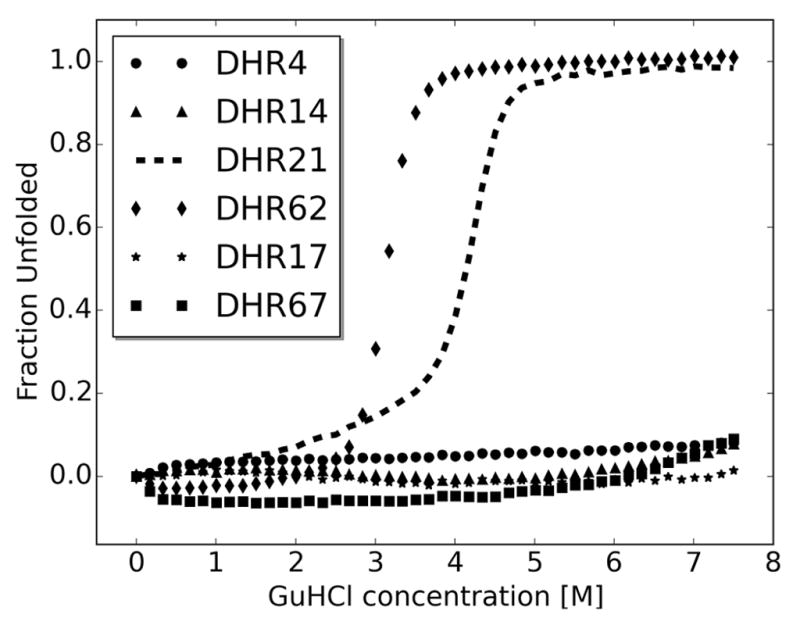
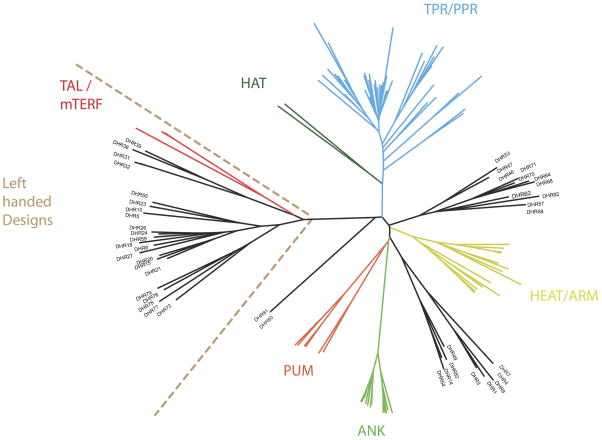
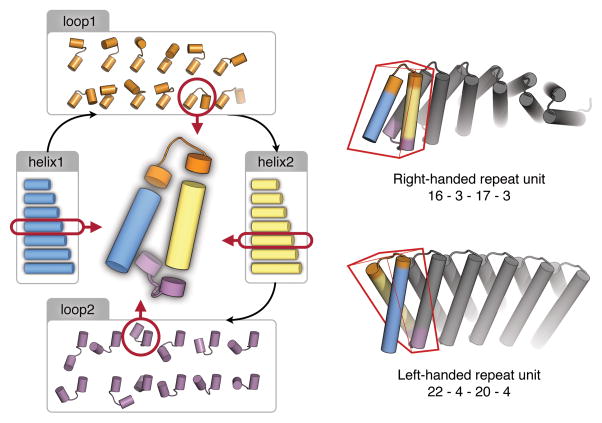
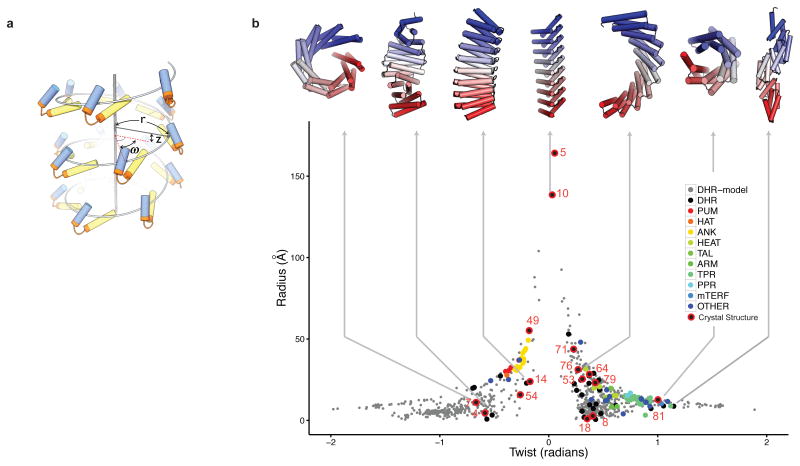
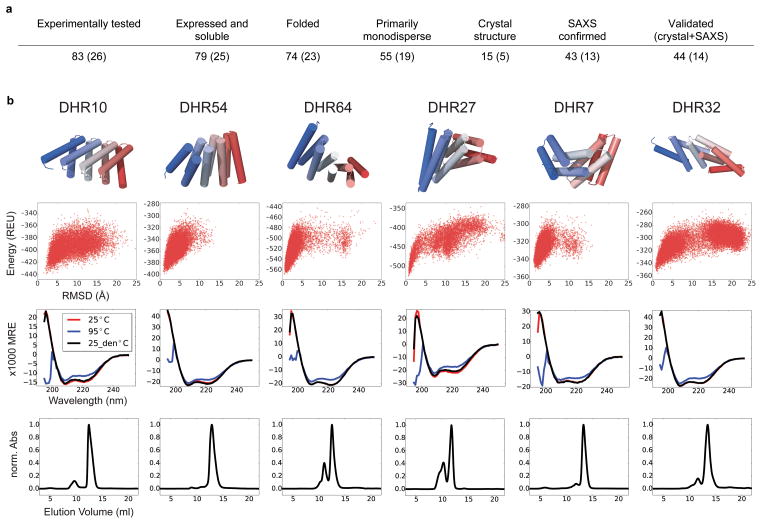
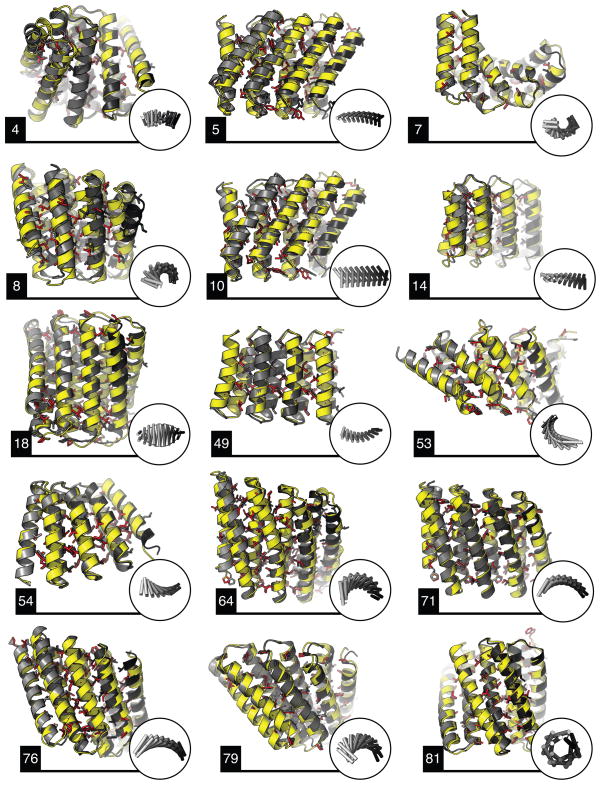
A central question in protein evolution is the extent to which naturally occurring proteins sample the space of folded structures accessible to the polypeptide chain. Repeat proteins composed of multiple tandem copies of a modular structure unit are widespread in nature and have critical roles in molecular recognition, signalling, and other essential biological processes. Naturally occurring repeat proteins have been re-engineered for molecular recognition and modular scaffolding applications. Here we use computational protein design to investigate the space of folded structures that can be generated by tandem repeating a simple helix-loop-helix-loop structural motif. Eighty-three designs with sequences unrelated to known repeat proteins were experimentally characterized. Of these, 53 are monomeric and stable at 95 °C, and 43 have solution X-ray scattering spectra consistent with the design models. Crystal structures of 15 designs spanning a broad range of curvatures are in close agreement with the design models with root mean square deviations ranging from 0.7 to 2.5 Å. Our results show that existing repeat proteins occupy only a small fraction of the possible repeat protein sequence and structure space and that it is possible to design novel repeat proteins with precisely specified geometries, opening up a wide array of new possibilities for biomolecular engineering.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
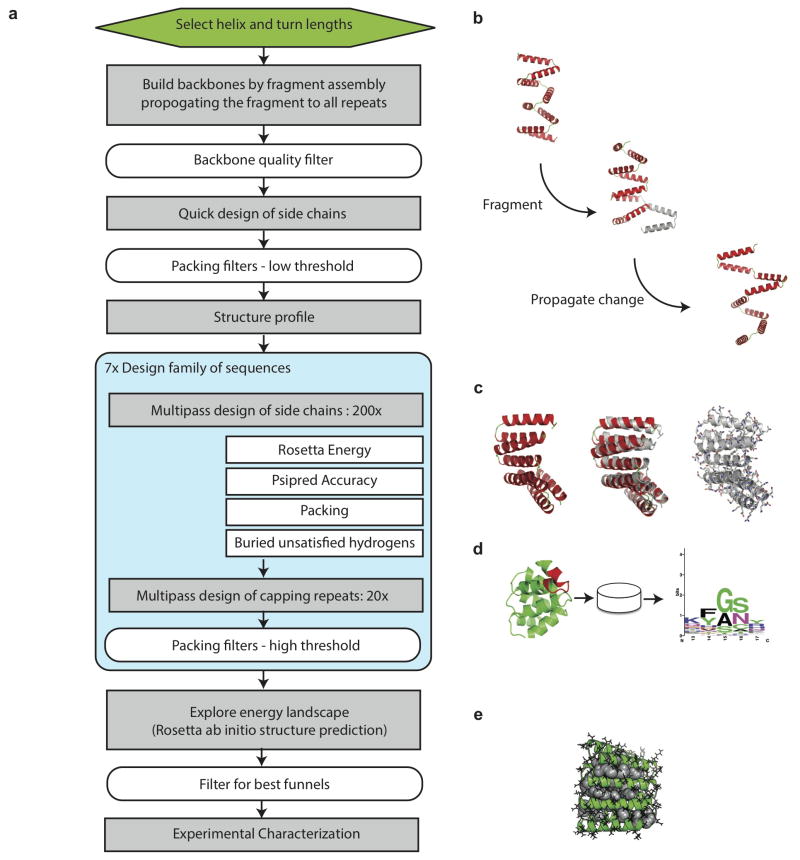
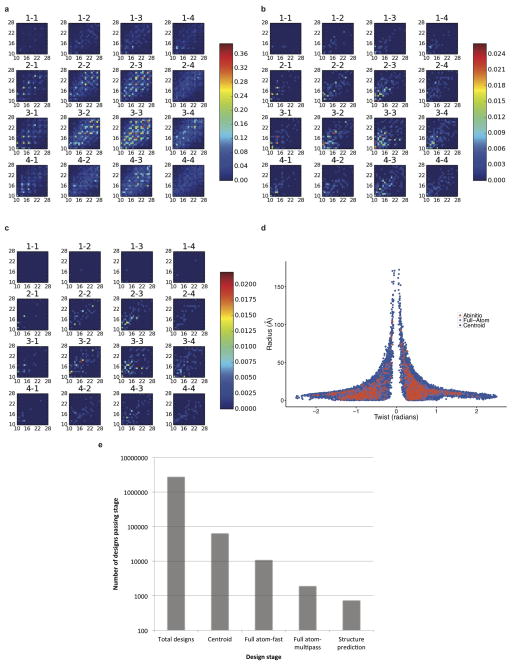
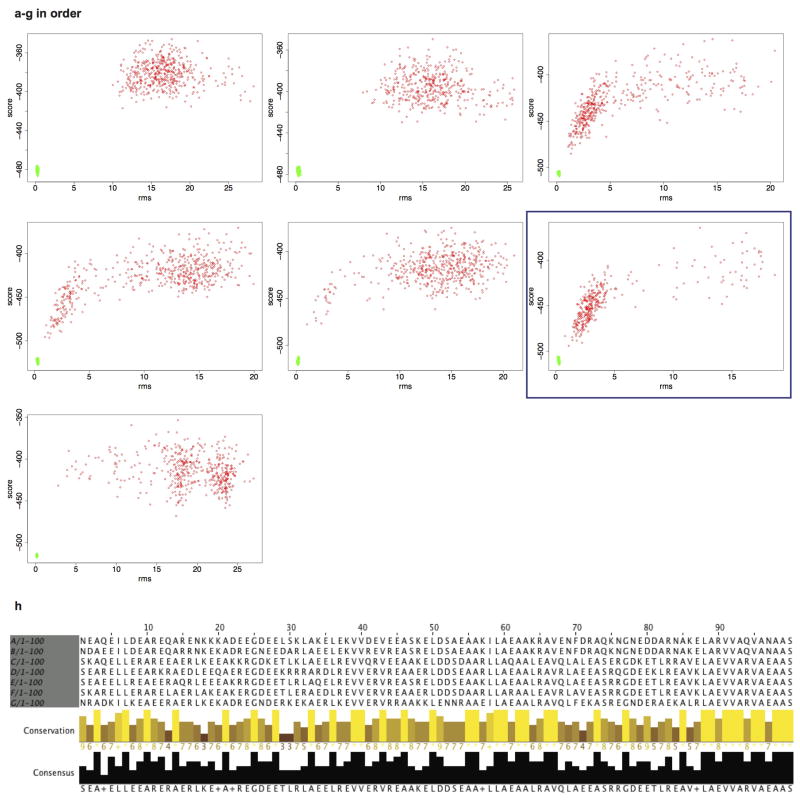
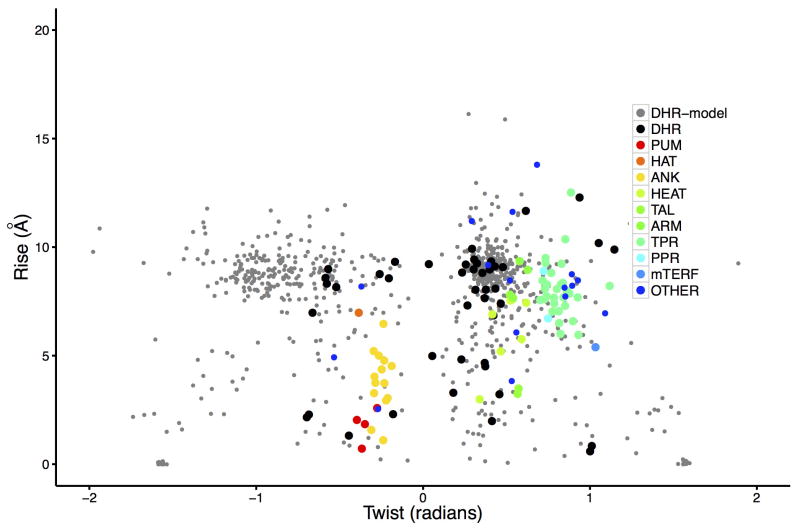


References
-
- Kajava AV. Tandem repeats in proteins: From sequence to structure. J Struct Biol. 2012;179:279–288. - PubMed
-
- Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. A census of protein repeats1. J Mol Biol. 1999;293:151–160. - PubMed
-
- Binz HK, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–582. - PubMed
-
- Varadamsetty G, Tremmel D, Hansen S, Parmeggiani F, Plückthun A. Designed Armadillo Repeat Proteins: Library Generation, Characterization and Selection of Peptide Binders with High Specificity. J Mol Biol. 2012;424:68–87. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
