

Structural and biochemical analysis of Escherichia coli ObgE, a central regulator of bacterial persistence
- PMID: 28223358
- PMCID: PMC5392579
- DOI: 10.1074/jbc.M116.761809
Structural and biochemical analysis of Escherichia coli ObgE, a central regulator of bacterial persistence
Abstract
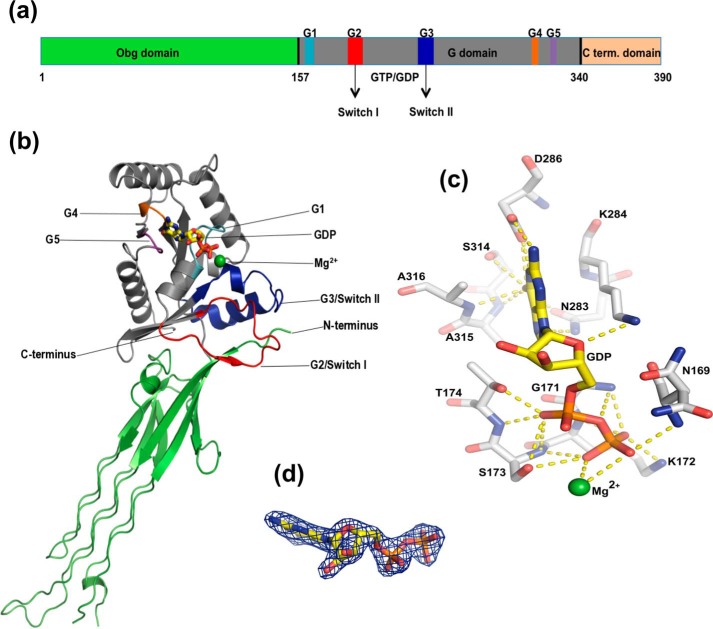
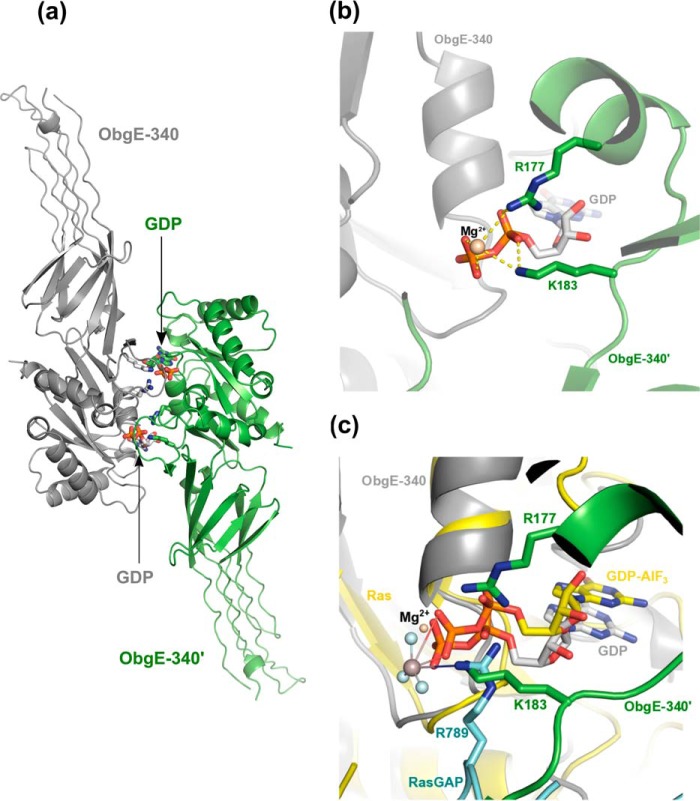
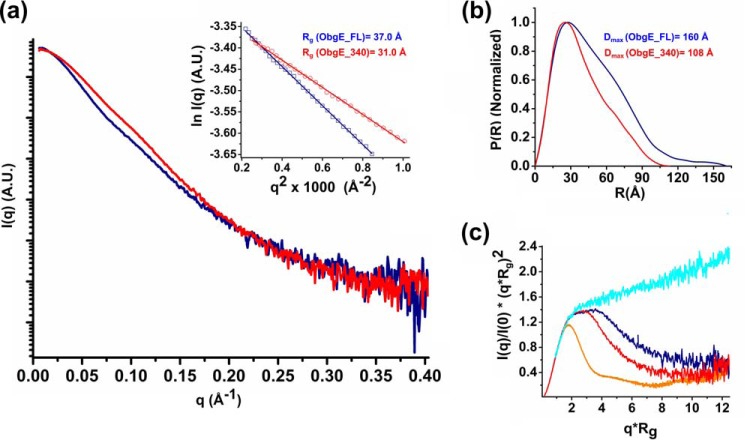
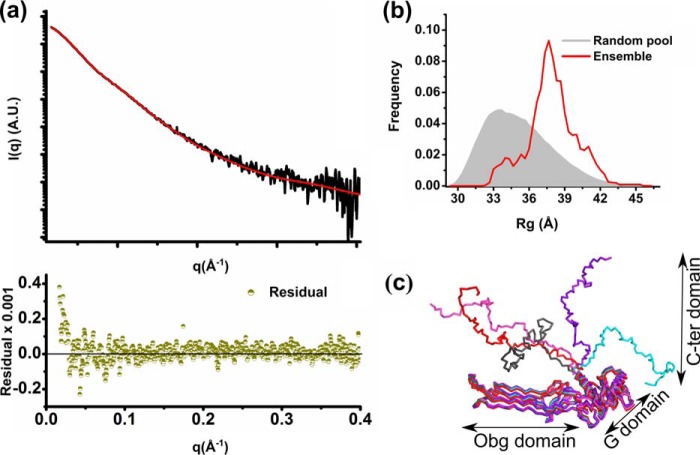
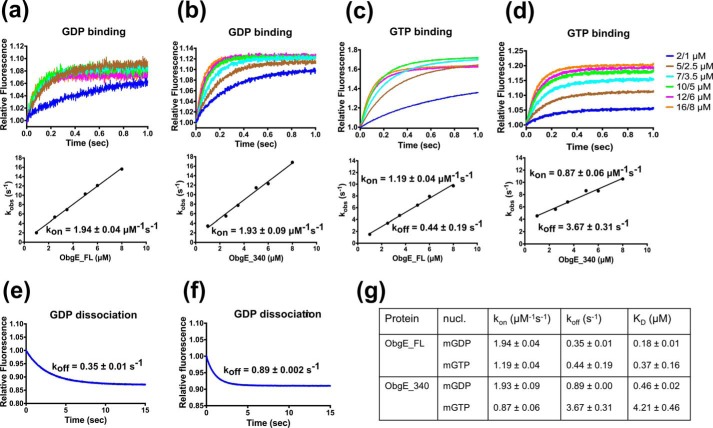
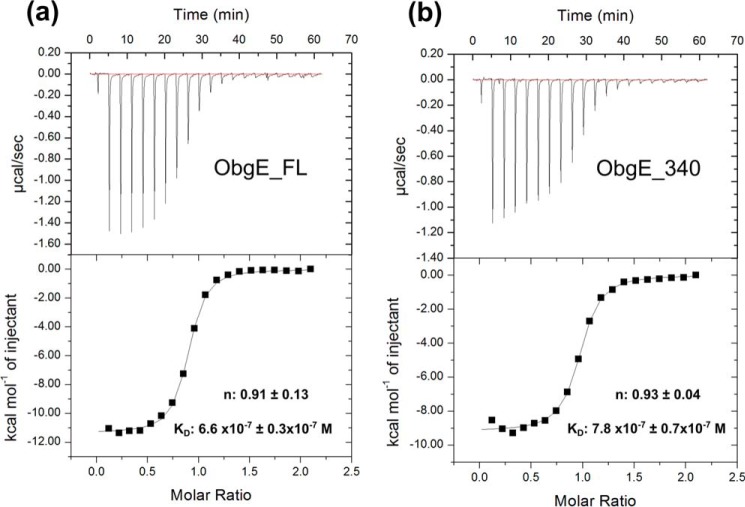
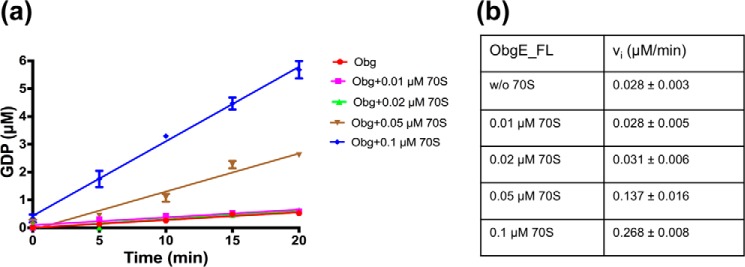
The Obg protein family belongs to the TRAFAC (translation factor) class of P-loop GTPases and is conserved from bacteria to eukaryotes. Essential roles in many different cellular processes have been suggested for the Obg protein from Escherichia coli (ObgE), and we recently showed that it is a central regulator of bacterial persistence. Here, we report the first crystal structure of ObgE at 1.85-Å resolution in the GDP-bound state, showing the characteristic N-terminal domain and a central G domain that are common to all Obg proteins. ObgE also contains an intrinsically disordered C-terminal domain, and we show here that this domain specifically contributed to GTP binding, whereas it did not influence GDP binding or GTP hydrolysis. Biophysical analysis, using small angle X-ray scattering and multi-angle light scattering experiments, revealed that ObgE is a monomer in solution, regardless of the bound nucleotide. In contrast to recent suggestions, our biochemical analyses further indicate that ObgE is neither activated by K+ ions nor by homodimerization. However, the ObgE GTPase activity was stimulated upon binding to the ribosome, confirming the ribosome-dependent GTPase activity of the Obg family. Combined, our data represent an important step toward further unraveling the detailed molecular mechanism of ObgE, which might pave the way to further studies into how this GTPase regulates bacterial physiology, including persistence.
Keywords: GTPase; X-ray crystallography; X-ray scattering; biophysics; crystal structure; enzyme kinetics; enzyme structure.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Bourne H. R., Sanders D. A., and McCormick F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132 - PubMed
-
- Caldon C. E., and March P. E. (2003) Function of the universally conserved bacterial GTPases. Curr. Opin. Microbiol. 6, 135–139 - PubMed
-
- Vetter I. R., and Wittinghofer A. (2001) The guanine nucleotide: binding switch in three dimensions. Science 294, 1299–1304 - PubMed
-
- Wittinghofer A., and Vetter I. R. (2011) Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 80, 943–971 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
