

Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence
- PMID: 32179646
- PMCID: PMC7196633
- DOI: 10.1074/jbc.RA120.012697
Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence
Abstract
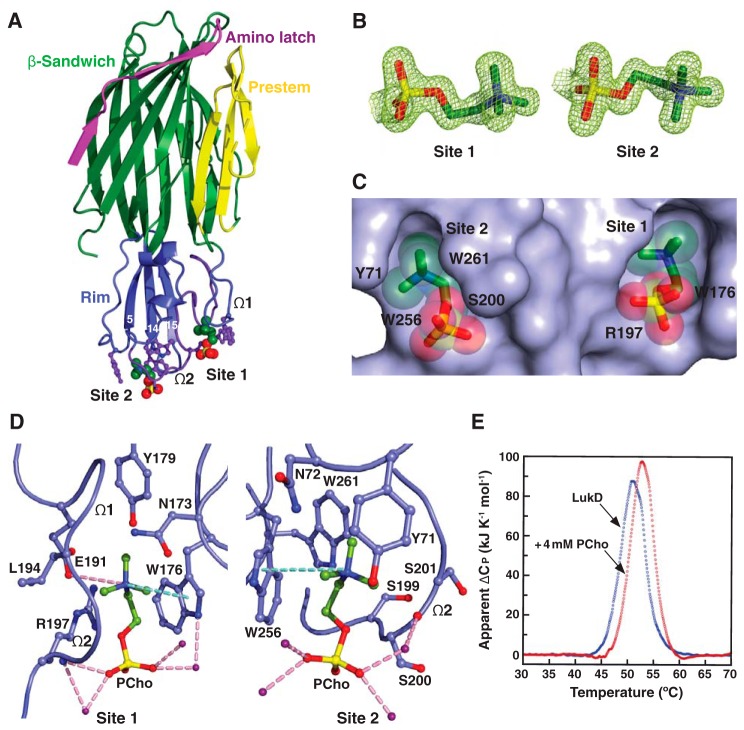
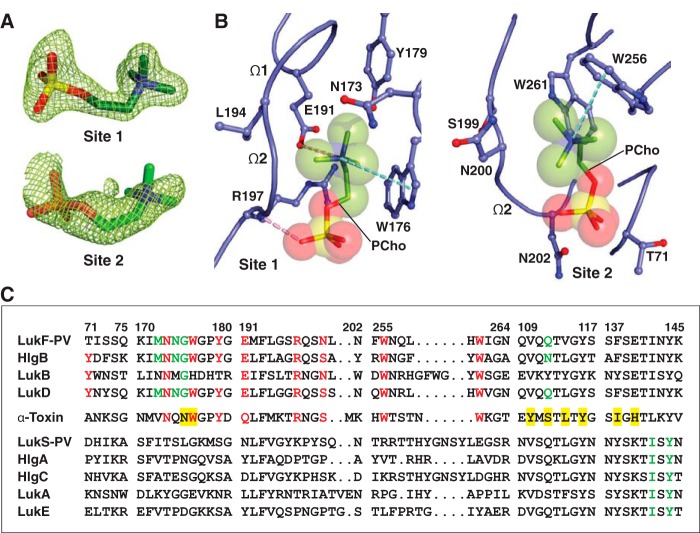
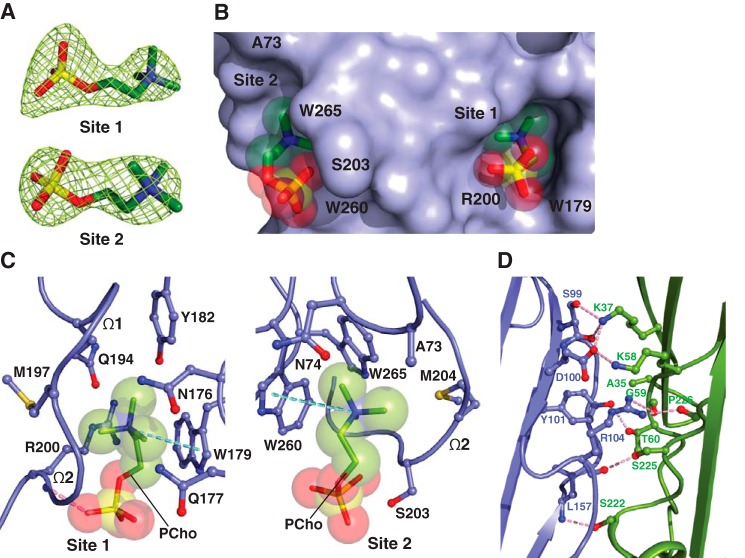
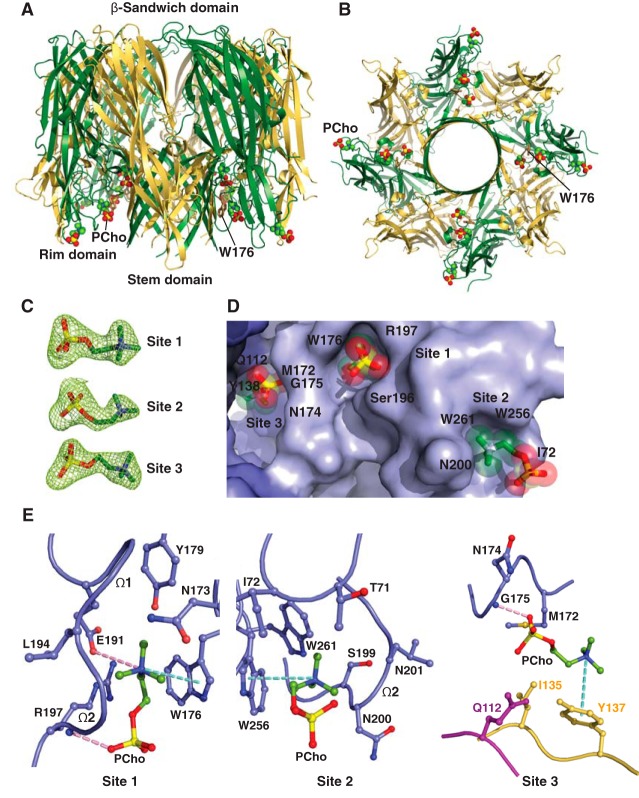
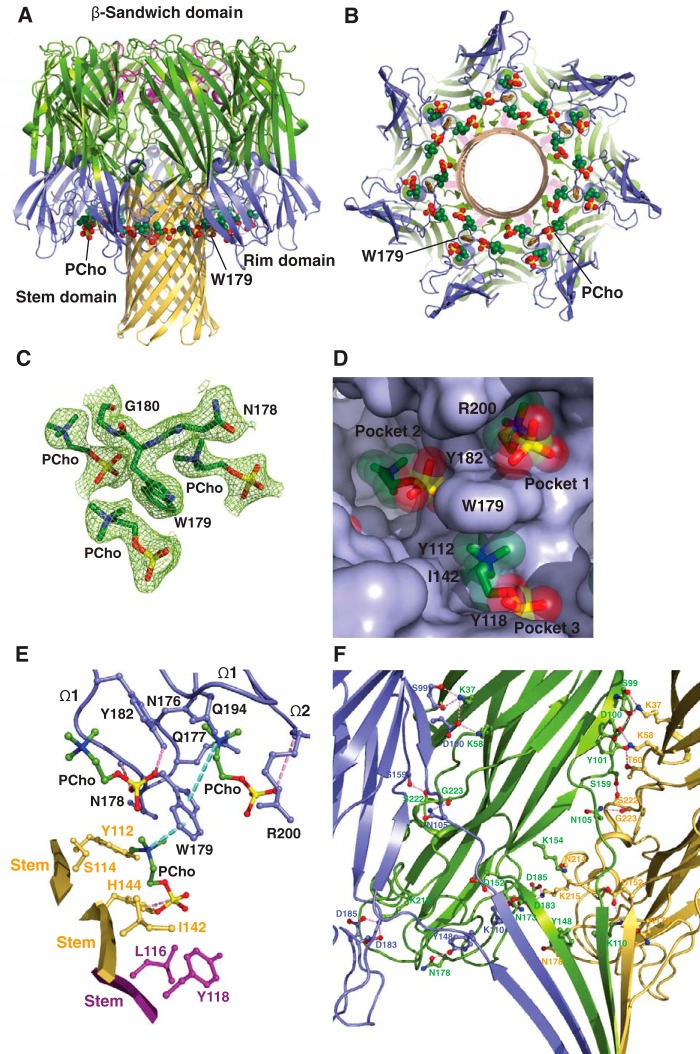
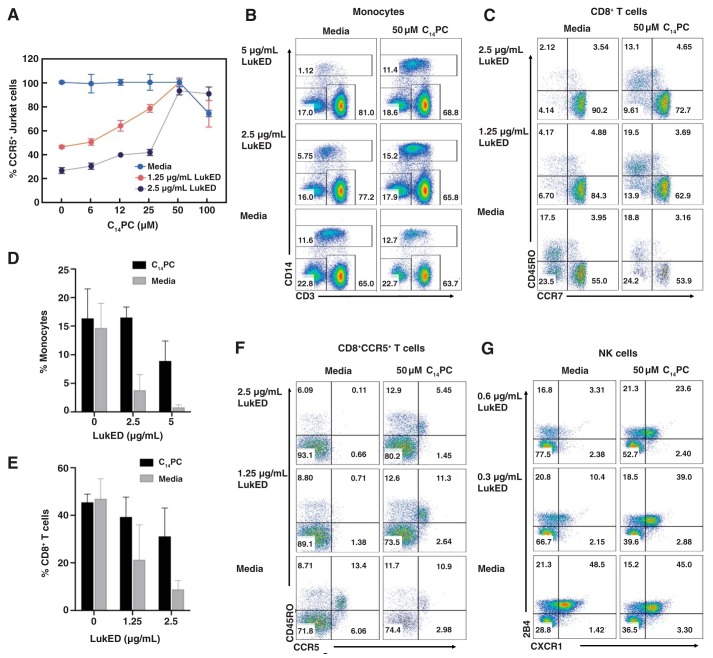
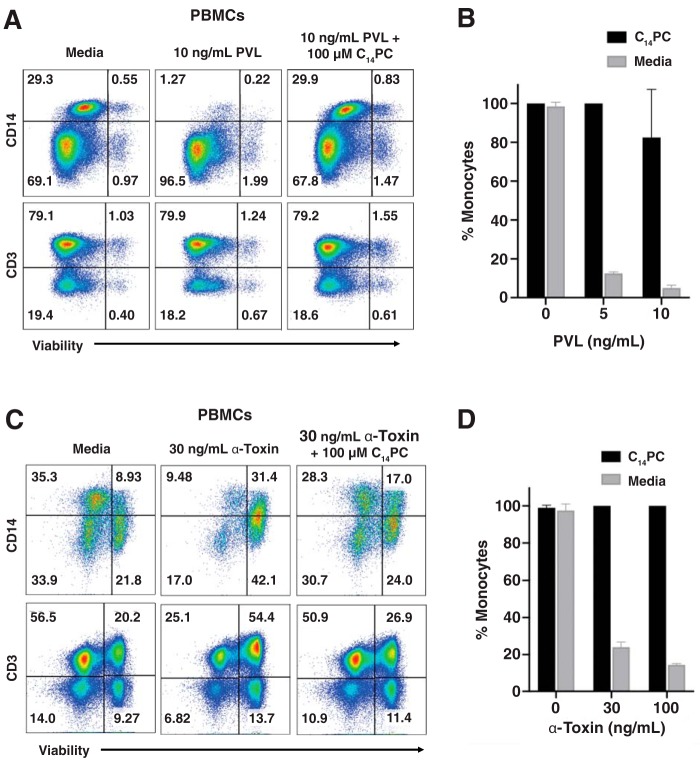
The rapid emergence and dissemination of methicillin-resistant Staphylococcus aureus (MRSA) strains poses a major threat to public health. MRSA possesses an arsenal of secreted host-damaging virulence factors that mediate pathogenicity and blunt immune defenses. Panton-Valentine leukocidin (PVL) and α-toxin are exotoxins that create lytic pores in the host cell membrane. They are recognized as being important for the development of invasive MRSA infections and are thus potential targets for antivirulence therapies. Here, we report the high-resolution X-ray crystal structures of both PVL and α-toxin in their soluble, monomeric, and oligomeric membrane-inserted pore states in complex with n-tetradecylphosphocholine (C14PC). The structures revealed two evolutionarily conserved phosphatidylcholine-binding mechanisms and their roles in modulating host cell attachment, oligomer assembly, and membrane perforation. Moreover, we demonstrate that the soluble C14PC compound protects primary human immune cells in vitro against cytolysis by PVL and α-toxin and hence may serve as the basis for the development of an antivirulence agent for managing MRSA infections.
Keywords: MRSA; Panton-Valentine leukocidin (PVL); alpha-toxin; antivirulence therapy; drug discovery; leukocidin ED (LukED); methicillin-resistant Staphylococcus aureus (MRSA); phosphatidylcholine; pore-forming toxin; structural biology; structure-based drug design; toxin; virulence factor.
© 2020 Liu et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Klevens R. M., Morrison M. A., Nadle J., Petit S., Gershman K., Ray S., Harrison L. H., Lynfield R., Dumyati G., Townes J. M., Craig A. S., Zell E. R., Fosheim G. E., McDougal L. K., Carey R. B., et al. (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771 10.1001/jama.298.15.1763 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
