

Molecular diagnoses in the congenital malformations caused by ciliopathies cohort of the 100,000 Genomes Project
- PMID: 34716235
- PMCID: PMC9340050
- DOI: 10.1136/jmedgenet-2021-108065
Molecular diagnoses in the congenital malformations caused by ciliopathies cohort of the 100,000 Genomes Project
Abstract
Background: Primary ciliopathies represent a group of inherited disorders due to defects in the primary cilium, the 'cell's antenna'. The 100,000 Genomes Project was launched in 2012 by Genomics England (GEL), recruiting National Health Service (NHS) patients with eligible rare diseases and cancer. Sequence data were linked to Human Phenotype Ontology (HPO) terms entered by recruiting clinicians.
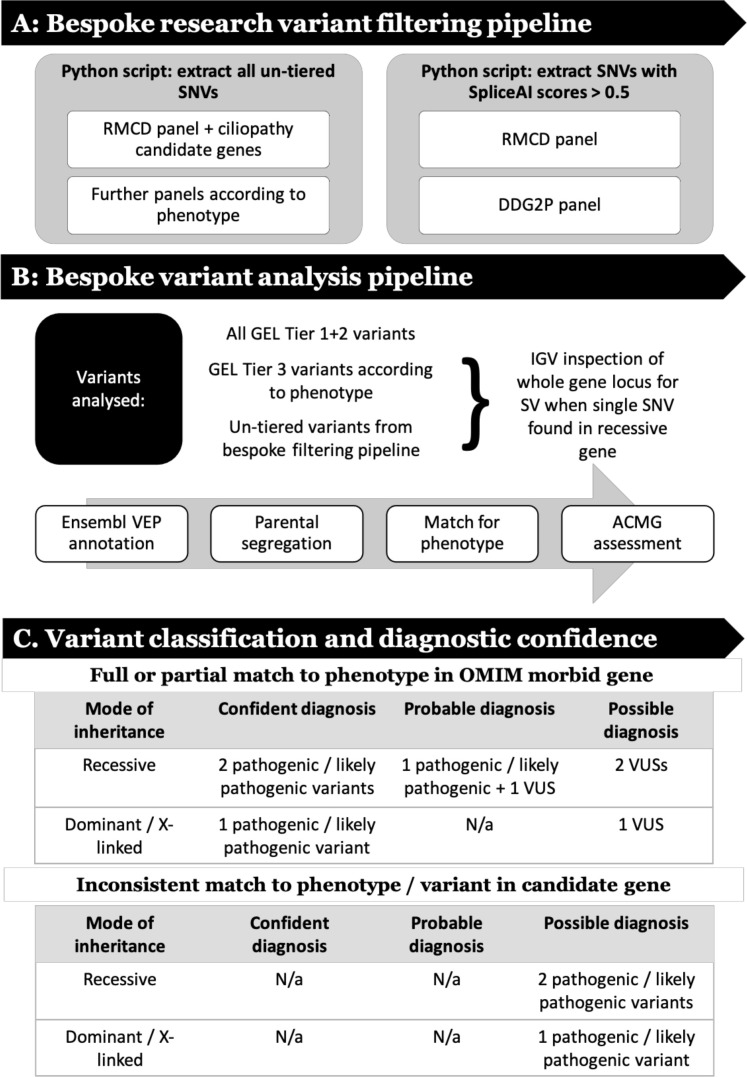
Methods: Eighty-three prescreened probands were recruited to the 100,000 Genomes Project suspected to have congenital malformations caused by ciliopathies in the following disease categories: Bardet-Biedl syndrome (n=45), Joubert syndrome (n=14) and 'Rare Multisystem Ciliopathy Disorders' (n=24). We implemented a bespoke variant filtering and analysis strategy to improve molecular diagnostic rates for these participants.
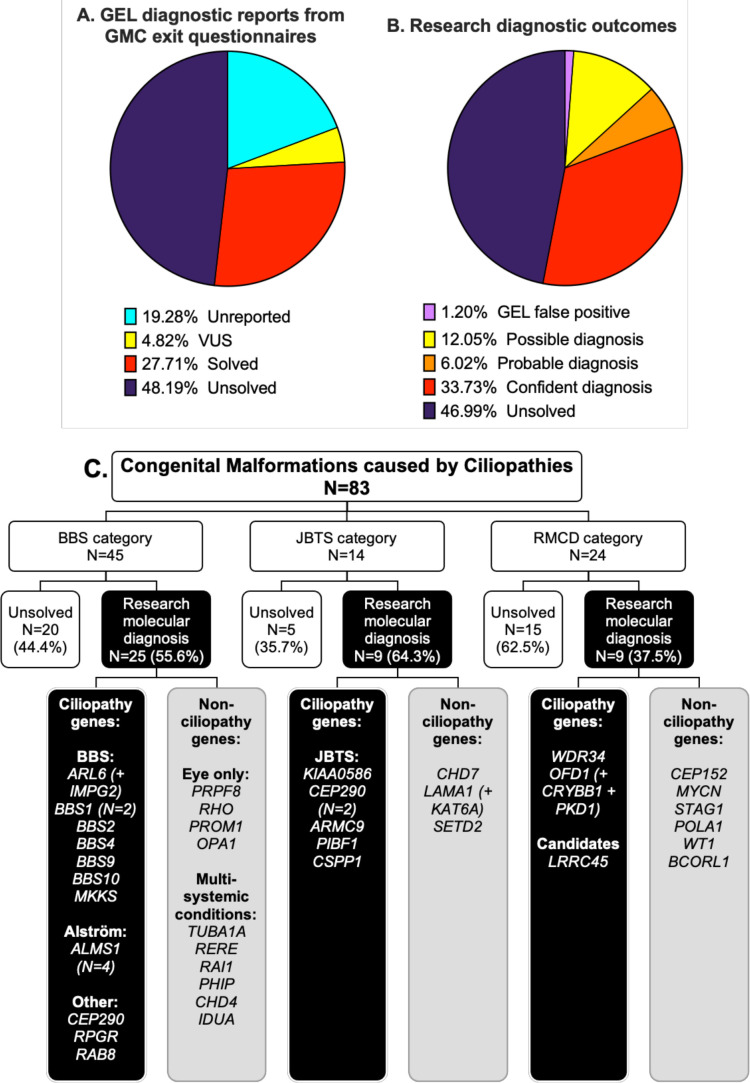
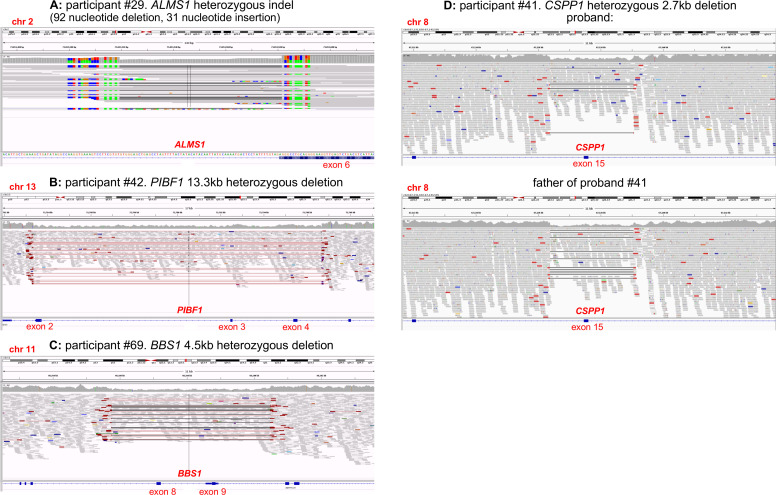
Results: We determined a research molecular diagnosis for n=43/83 (51.8%) probands. This is 19.3% higher than previously reported by GEL (n=27/83 (32.5%)). A high proportion of diagnoses are due to variants in non-ciliopathy disease genes (n=19/43, 44.2%) which may reflect difficulties in clinical recognition of ciliopathies. n=11/83 probands (13.3%) had at least one causative variant outside the tiers 1 and 2 variant prioritisation categories (GEL's automated triaging procedure), which would not be reviewed in standard 100,000 Genomes Project diagnostic strategies. These include four structural variants and three predicted to cause non-canonical splicing defects. Two unrelated participants have biallelic likely pathogenic variants in LRRC45, a putative novel ciliopathy disease gene.
Conclusion: These data illustrate the power of linking large-scale genome sequence to phenotype information. They demonstrate the value of research collaborations in order to maximise interpretation of genomic data.
Keywords: and neonatal diseases and abnormalities; congenital; diagnosis; genetics; genomics; hereditary; medical.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: Disclosure: HB, RPJB and AS are employed by Genomics England, UK. GW is employed by Illumina. The other authors declare no conflict of interest.
Figures
References
-
- Barsch F, Niedermair T, Mamilos A, Schmitt VH, Grevenstein D, Babel M, Burgoyne T, Shoemark A, Brochhausen C. Physiological and pathophysiological aspects of primary Cilia-A literature review with view on functional and structural relationships in cartilage. Int J Mol Sci 2020;21:4959. 10.3390/ijms21144959 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical